Difference between revisions of "Metachromatic leukodystrophy 2011"
| Line 78: | Line 78: | ||
* TASK 2: [[metachromatic_leukodystrophy_reference_aminoacids|Alignments]] |
* TASK 2: [[metachromatic_leukodystrophy_reference_aminoacids|Alignments]] |
||
* TASK 3: [[Sequence-based analyses of ARS A|Sequence-based analyses of ARS A]] |
* TASK 3: [[Sequence-based analyses of ARS A|Sequence-based analyses of ARS A]] |
||
| + | * TASK 4: [[Homology Modeling of ARS A|Homology Modeling of ARS A]] |
||
== References == |
== References == |
||
Revision as of 11:56, 7 June 2011
Contents
Summary
Metachromatic leukodystrophy (MLD) is an incurable, autosomal recessive inherited disease, which is caused by a mutation in the enzyme Arylsulfatase A (ARS A). It belongs to the lysosomal storage diseases. The deficiency of the enzyme leads to storage of sulfatides and lysosulfatides, which are usually digested by the enzyme. The storage of the lipids glycosphingolipid sulfatide and lysosulfatide cause a degeneration of the white matter in the brain. This part of the brain mainly consists of myelinated axons, which are needed for the proper function of the brain. <ref name="blomqusit2011">Blomqvist, Maria. Gieselmann,Volkmar. Mansson,Jan-Eric. "Accumulation of lysosulfatide in the brain of arylsulfatase A-deficient mice". Lipids in Health and Disease, 2011</ref> Sulfatide is one of many compomnents of the complex composition of myelin <ref name="jeon2008">Jeon, S. B. and Yoon, H. J. and Park, S. H. and Kim, I. H. and Park, E. J.. "Sulfatide, a major lipid component of myelin sheath, activates inflammatory responses as an endogenous stimulator in brain-resident immune cells. J Immunol, 2008</ref>. ARS A catalyses the breakdown and correct incorporation <ref name="wiesmann1978">Wiesmann, U. N.. "[Pathophysiology of sulfatide metabolism in metachromatic leukodystrophy]". Bull Schweiz Akad Med Wiss, 1978</ref> There is no effective therapy available yet and carries of the disease all die. The progression and symptoms of the disease are in general mental and motoric retardation. <ref name="biffi:2008">Biffi, A. and Lucchini, G. and Rovelli, A. and Sessa, M. "Metachromatic leukodystrophy: an overview of current and prospective treatments." Bone Marrow Transplant ,2008 </ref>
Phenotype
Five different allelic variants have been described. Depending on the variant, onset and type of symptoms and progression of the disease can be different <ref name="wikipedia"> http://en.wikipedia.org/wiki/Metachromatic_leukodystrophy </ref>, <ref name="OMIM"> http://www.ncbi.nlm.nih.gov/omim/250100 </ref>:
- Late infantile: Carriers of the disease causing alleles usually become diseased two years after birth. The patients show motor symptoms, rigidity, mental deterioration, and sometimes convulsions. Death occurs five years after breakout of the disease at the latest.
- Juvenile: Onset is between 3 to 10 years of age. The disease usually begins with impaired school performance, mental deterioration and dementia. Then, symptoms strongly resemble the late infantile form. As the progression is slower than in the juvenile form, patients normally die 10 to 15 years after the start of the symptoms.
- Adult forms: This form of MLD begins after the age of 16. Symptoms are commonly psychiatric and can lead to the diagnosis of schizophrenia. Disorders in movement and posture appear very late. The progression of the disease is even slower than in the "late infantile" and "juvenile" forms and patients may live for another several decades after onset of the symptoms.
- Partial cerebroside sulfate deficiency: Carriers of this allele show apparent ARS A enzyme deficiency, but without neurologic abnormalities.
- Pseudoarylsulfatase A deficiency: In this form, the enzyme is partial defected with 10-20 % of normal activity. The symptoms are similar to adult form.
Cross-references
See also description of this disease in
- Wikipedia article on MLD
- Wikipedia article on ARS A
- HGMD entry of ARS A
- OMIM entry for MLD
- OMIM Entry for ARS A
- KEGG disease entry for MLD
Biochemical disease mechanism
Arylsulfatase A is located in the Lysosome, where it is responsible for breaking up sulfated Glycosphingolipids (Sulfatides). As the sulfatides are not degraded, they build up and destroy the myelin sheath around the axons.

Cross-references
Genomic and structural basis of Arylsulfatase A
Gene
The gene coding for Arylsulfatase A is the ARSA gene. It is located on the long arm of chromosome 22 base pair 51,063,448 to base pair 51,066,606. It consists of eight exons and is 2.3 kb long. <ref name="biffi:2008">Biffi, A. and Lucchini, G. and Rovelli, A. and Sessa, M. "Metachromatic leukodystrophy: an overview of current and prospective treatments." Bone Marrow Transplant ,2008 </ref>

Function
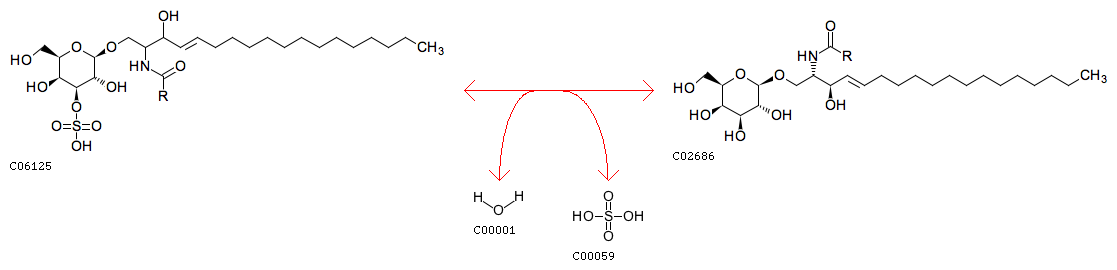
Arylsulfatase A is an enzyme in the lysosome that catalyses the break-up of sulfatides into galactosylceramides and sulfates by acting on the ester bonds.

{kind=link}
Structure
Arylsulfatase A consists of two subunits A1 and A2 which are connected by a short linker. Both subunits contain a nucleotide-binding-site and they are both needed for proper function of the enzyme. <ref name="zhou:2000">Tongqing Zhou, Sergei Radaev, Barry P. Rosen and Domenico L. Gatti "Structure of the ArsA ATPase: the catalytic subunit of a heavy metal resistance pump" The EMBO Journal ,2000 </ref>

Cross-references
Treatment
There is no effective therapy avalable yet. Efforts are made to develop and test promising medication for the future <ref name="biffi:2008">Biffi, A. and Lucchini, G. and Rovelli, A. and Sessa, M. "Metachromatic leukodystrophy: an overview of current and prospective treatments." Bone Marrow Transplant ,2008 </ref>:
- Enzyme replacement therapy: The deficient enzyme is replaced, e.g. by infusion and thus restores enzyme activity. Currently this is the most promising approach, because it is already available for the following lysosomal storage diseases: Gaucher disease, Fabry disease, MPS I, MPS VI and Glycogen storage disease type II. Clinical tests of this approach are conducted at the moment.
- Gene therapy: Gene therapy may be used to e.g. repopulate affected tissues with donor-derived myeloid cells. Also here, clinical test are currently performed.
Mutations
47 known, disease causing allelic variants are listed in the OMIM database.
Links to mutated sequences are coming soon.
Reference sequence
The reference sequence does not cause the disease and is most often found in the population.
TASKS
- TASK 2: Alignments
- TASK 3: Sequence-based analyses of ARS A
- TASK 4: Homology Modeling of ARS A
References
<references />