Difference between revisions of "Hemochromatosis"
| Line 40: | Line 40: | ||
| + | == Pathophysiology == |
||
| − | == Biochemical disease mechanism == |
||
=== The role of iron in the body === |
=== The role of iron in the body === |
||
| Line 63: | Line 63: | ||
In the most common variant of hemochromatosis, a point mutation in the HFE gene disrupts the hepcidin production pathway. The iron resorption is increased and it accumulates in the body. At some point the body cannot store more iron in the cells and Fe<sup>2+</sup> occurs unbound in the bloodstream which damages the body. |
In the most common variant of hemochromatosis, a point mutation in the HFE gene disrupts the hepcidin production pathway. The iron resorption is increased and it accumulates in the body. At some point the body cannot store more iron in the cells and Fe<sup>2+</sup> occurs unbound in the bloodstream which damages the body. |
||
| − | + | Thus, patients do not suffer from a perturbance of the iron metabolism, which works normal, but from an increased iron uptake into the blood. |
|
Revision as of 22:18, 28 April 2013
Work in progress!
{kind=link}
Hemochromatosis is a hereditary disorder that leads to an increased intestinal iron uptake from food. The excess iron is stored in the parenchymal cells of organs and tissues and thus disrupts their normal function. There are various genotypes that can lead to hemochromatosis. The most common form of the disease is caused by a point mutation in the HFE gene. HFE stands for High FErrum.
Contents
Phenotype
Clinical symptoms include:
- tiredness
- joint and bone pain
- destructive arthritis
- liver fibrosis and cirrhosis, increased risk to develop hepatocellular carcinoma
- Glucose intolerance and insulin resistance due to damages in the pancreas (diabetes mellitus).
- Gonadal dysfunction, hypogonadism and decreased libido.
- heart failure, arrhythmias or pericarditisheart failure
- grey or dark cutaneous (skin) pigmentation
The hemocromatosis phenotype and its harmfulness varies in patients with the same disease causing mutation. Symptoms are caused by a high, toxic iron accumulation in parenchymal cells of important organs, like the heart, the liver or the endocrine glands.
The disease is usually diagnosed in middle aged patients, because the iron accumulates over time and severe symptoms are not immidiately apparent.
Depending on which organs are affected most by the increased iron uptake, the symptoms range from simple biochemical abnormalities to severe diseases such as heart failure and liver cirrhosis.
The individual phenotype varies that much, because the genetic background only gives a predisposition to hemochromatosis. Human and environmental factors play an important role as well. In general, male indiviuals that have the genetic predisposition, have a higher probability of developing hemochromatosis, supposedly because females have an overall higher iron loss due to menstruation.
Cross references
see also the description in
http://en.wikipedia.org/wiki/Hereditary_haemochromatosis
http://omim.org/entry/235200
http://www.gastrojournal.org/article/S0016-5085%2810%2900872-3/fulltext
Pathophysiology
The role of iron in the body
Iron is the 4th most common element in the world. In nature, it normally occurs in its oxidized form Fe3+, that is unsoluble in water. That is, why iron in humans mostly occurs complexed. Free, water soluble iron ions that appear in the reduced form Fe2+, are toxic because they can react with H2O2 to produce highly reactive hydroxy radicals that damage membranes, proteins and nucleic acids. Nevertheless, iron plays a crucial role in the human body. It binds to hemoglobin as a cofactor and enables oxygen transport. The total amount of iron in the body ranges from 3000mg-5000mg, whereby around 65% are bound to hemoglobin and the rest is either bound to other proteins as cofactor or stored in cells as reservoir. Iron is only removed from the body through blood loss, sweating or shedding of mucosal or skin cells. This amounts to an average of 1 mg per day for men and 1.5-2 mg per day for women. Women lose more iron, because they regularly loose blood due to menstruation. The amount of iron in a healthy human is kept constant by the regulatory mechanisms of the body. This means that only as much, as the organism looses, is taken up.

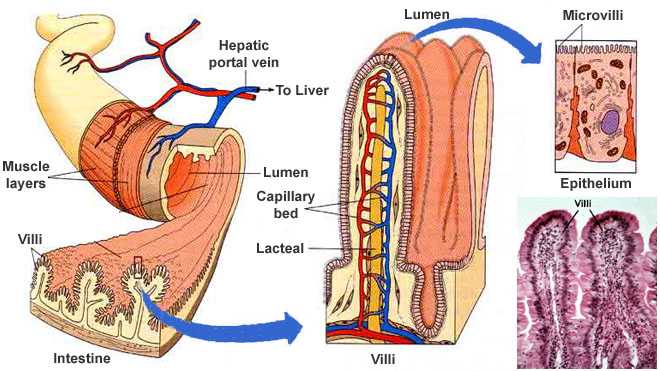{kind=link}
Iron is only able to enter the human body in the small intestine. The main part is taken up in the duodenum and the resorbtion decreases linearly towards the ileum(end of the small intestine). The iron in the lumen of the small intestine is transported into the enterocytes through the divalent metal transporter(DMT-1). There, the iron is either stored inside the cell in complex with the ferritin protein or it is released into the bloodstream via ferroportin(FPN-1), where it immediately forms a complex with transportin. The amount of iron that enters the bloodstream highly depends on the expression level of FPN-1. The enterocytes are regularly depleted and thus the iron that they have stored is lost.
The iron-transferrin complex in the blood plasma binds to the Transferrin Receptors 1 and 2(TfR) that the parenchymal cells of the liver express. These receptors start a signalling pathway involving the HFE-protein that activates the production of hepcidin. Although there are some theories about this pathway, it is not resolved yet. Hepcidin is a protein that is responsible for the downregulation of iron entry into the bloodstream, because it initiates the degradation of FPN-1. Therefore, the release of iron into the blood plasma is decreased.

{kind=link}
Disease Mechanism
In the most common variant of hemochromatosis, a point mutation in the HFE gene disrupts the hepcidin production pathway. The iron resorption is increased and it accumulates in the body. At some point the body cannot store more iron in the cells and Fe2+ occurs unbound in the bloodstream which damages the body.
Thus, patients do not suffer from a perturbance of the iron metabolism, which works normal, but from an increased iron uptake into the blood.
Genetics and Inheritance
There are several different types of hemochromatosis. Each type is connected to defects in the iron uptake regulation through hepcdin. The most common and less severe type is caused by a mutation in the HFE gene on chromosome 6. The other types are rare and based on a mutations in the TfR2 gene or, in the case of juvenile hemochromatisis, mutations in the HJV or HAMP (Hepcidin) gene. Mutations in the FPN(Ferroportin) gene can also result in hemachromatosis like symptoms, but it is often termed ferroportin disease. The mutations leads to a hepcidin resistance and thus to an iron hyperabsorbtion from the diet, although the hepcidin production is not impaired in this patients.
Homozygosity for one of the above mentioned mutations only results in a certain predisposition to hemochromatosis, but not all persons with this genetic background develop the disease. It depends on other influences on the iron metabolism, such as abusive drinking.
| form | male/female | age | non-/caucasian |
|---|---|---|---|
| HFE | male | 40-50 years | caucasian |
| TfR2 | male or female | 30-40 years | caucasian or non-caucasian |
| HJV, HAMP | male or female | 15-20 years | caucasian or non-caucasian |
| Ferroportin disease | male or female | 10-80 years | caucasian or non-caucasian |
Hemochromatosis is an autosomal recessive inherited disorder with reduced penetrance in female. The HFE C282Y mutation is a very common one with a homozygozity prevalence of 1:200 to 1:300 in white persons, why it is a polymorphism rather than a disease mutation. But among Asians, Hispanics or black persons it is much less common. Roughly 80% of northern European hemachromatosis patients are homozygous for HFE C282Y. A Celtic or Vikin ancester was probably the first person having the mutation. Since hemochromatisis does not infect reproduction, the mutation spread through populations. The C282Y allel has a frequency of 12.5% in Ireland and 0% in southern Europe, but the mean frequency among white individuals is 6%.

H63D is anothe HFE polymorphism with less geographic distribution and a higher prevalence of 14%, but it has nearly no penetrance. The S65C polymorphism is associated with high iron levels when it is inherited together with C282Y on one allele.
All the other non-HFE hemochromatosis types are spread throughout the world, independent of any race, but they are much rarer. In 50% of families with a case of juvenile hemachromatose has the G320V mutation in the HJV gene been detected.
This wiki entry focuses on the HFE C282Y mutation, because it is the most common reason for hemachromatosis in northern Europe.
HFE C282Y
The HFE gene is located on chromosome 6 on the short arm (p) in region 21.3 (6p21.3) and consists of 7 exons spanning 12 kb. It encodes for the HFE protein, which is a membrane protein similar to the MHC class I proteins and HFE wthuw associates with beta2-microglobulin. HFE binds to the transferrin receptor 2 (TfR2) and is thought to play a role in the pathway that senses the serum iron level (transferrin-bound and un-unbound) and regulates the expression of hepcidin.
A guanin to adenine transition at position 845 in the HFE gene lead to the C282Y substitiution in the protein sequence. This missense mutation affects a highly conserved cystein in the alpha-3 loop of the HFE protein that normally forms a disulfide bond in the unmutated protein.(see picture above) The tyrosine in the mutated protein therefore disrupts the structure and prevents the binding of HFE to beta-2-microglobulin.
Cross references
see also the description in
http://omim.org/entry/613609
http://omim.org/entry/235200
http://www.genecards.org/cgi-bin/carddisp.pl?gene=HFE&search=HFE&suff=txt
Mutations
Each of the above mentioned different types of hemochromatosis is caused by a different mutation.
HFE: C282Y, H63D
TfR2: Y250X (nonsense mutation, it truncates TfR2 at amoino acid 250)
HJV: G320V
HAMP: several, no specific
FPN: C326S and C326Y
Reference sequence
protein sequence:
>gi|1890180|emb|CAB07442.1| HFE [Homo sapiens] MGPRARPALLLLMLLQTAVLQGRLLRSHSLHYLFMGASEQDLGLSLFEALGYVDDQLFVFYDHESRRVEP RTPWVSSRISSQMWLQLSQSLKGWDHMFTVDFWTIMENHNHSKESHTLQVILGCEMQEDNSTEGYWKYGY DGQDHLEFCPDTLDWRAAEPRAWPTKLEWERHKIRARQNRAYLERDCPAQLQQLLELGRGVLDQQVPPLV KVTHHVTSSVTTLRCRALNYYPQNITMKWLKDKQPMDAKEFEPKDVLPNGDGTYQGWITLAVPPGEEQRY TCQVEHPGLDQPLIVIWEPSPSGTLVIGVISGIAVFVVILFIGILFIILRKRQGSRGAMGHYVLAERE
http://www.ncbi.nlm.nih.gov/protein/1890180?report=fasta
Diagnosis and Treatment

Hemochromatosis is diagnosed in patients with an unnormal high transferrin saturation (TS) and, in later stages, increased serum ferritin levels. The transferrin saturation denotes the concentration of free iron in proportion to the concentration of transferin in the blood serum. However, the diagnosis should always be supported by a gene test for HFE C282Y homyozygocity. Inflammation, metabolic disorders, diabetes mellitus, alcohol abuse and liver cell necrosis can also lead to an increased serum ferritin level. On the other hand, a finding of normal serum ferritin level alsways excludes hemochromatosis.
Since iron can only be removed from the system by blood loss, the only possible treatment is phlebotomy (bloodletting). It is aimed to reduce the iron content in the body. The first step of the iron-depletion treatment is to induce a slightly iron-deficient state in the body. Therfore, 400-500 ml blood are removed weekly. After 1 to 2 years a serum ferrition level of 20-50 μg/l is reached. In the long term, a maintenance therapy with two to four phlebotomies a year is then enough to keep the serum ferritin level between 50-100 μg/l.
A low iron diet can help. The life expectancy of dignosed and treated hemochromatosis patients withour complications is comparable to that of the normal population. But early diagnosis and initation of therapy increase survival time.
Resources
The entry is based on several resources:
http://www.gastrojournal.org/article/S0016-5085%2810%2900872-3/fulltext
http://omim.org/entry/613609
http://omim.org/entry/235200
http://en.wikipedia.org/wiki/Hereditary_haemochromatosis
http://www.genecards.org/cgi-bin/carddisp.pl?gene=HFE&search=HFE&suff=txt