Difference between revisions of "Task 4 (MSUD)"
(→Structural alignments) |
m (→Structural alignments) |
||
| Line 130: | Line 130: | ||
==== Structural alignments ==== |
==== Structural alignments ==== |
||
| − | <gallery perrow=2 widths=400px heights= |
+ | <gallery perrow=2 widths=400px heights=250px caption="Structures superimposed to X-ray structures of BCKDHA"> |
File:Bckdha-aligned-to-2bfe.png|BCKDHA aligned to 2BFE. Structure of BCKDHA is shown in green. |
File:Bckdha-aligned-to-2bfe.png|BCKDHA aligned to 2BFE. Structure of BCKDHA is shown in green. |
||
File:Bckdha-aligned-to-3exg.png|BCKDHA aligned to 3EXG. Structure of BCKDHA is shown in green. |
File:Bckdha-aligned-to-3exg.png|BCKDHA aligned to 3EXG. Structure of BCKDHA is shown in green. |
||
Revision as of 15:34, 3 June 2013
Contents
Structural alignment
Results
Following are the structures we have chosen for evaluation of structural alignment methods.
|
PDB ID |
Identity to BCKDHA |
Origin |
Comment |
|
1u5b |
100% |
Mitochondrial branched-chain α-keto acid dehydrogenase (Human) |
BCKDHA |
|
2bfe |
99% |
Mitochondrial branched-chain α-keto acid dehydrogenase (Human) |
BCKDHA alternative structure |
|
3exg |
24.9% |
Pyruvate dehydrogenase (Human) |
structure with low identity |
|
13pk |
4.1% |
Phosphoglycerate kinase (Trypanosoma brucei) |
same CAT classification |
|
17gz |
8.3% |
Glutathione S-Transferase (Human) |
same CA classification |
|
2z37 |
5.4% |
Chitinase (Brassica juncea) |
same C classification |
|
1f0y |
8.1% |
L-3-Hydroxyacyl-CoA Dehydrogenase (Human) |
different CATH classification |
We did not find PDB structure which has a sequence identity between 40% to 90% in comparison to BCKDHA.
Structural alignments
- Structures superimposed to X-ray structures of BCKDHA
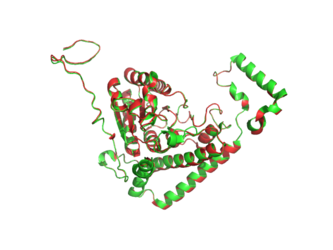
BCKDHA aligned to 2BFE. Structure of BCKDHA is shown in green.
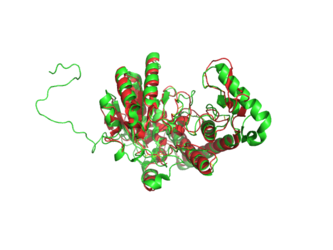
BCKDHA aligned to 3EXG. Structure of BCKDHA is shown in green.
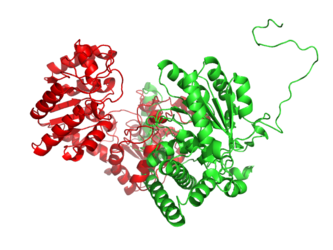
BCKDHA aligned to 13PK. Structure of BCKDHA is shown in green.

BCKDHA aligned to 17GS. Structure of BCKDHA is shown in green.

BCKDHA aligned to 2Z37. Structure of BCKDHA is shown in green.
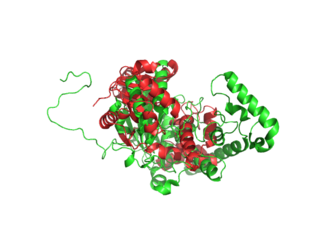
BCKDHA aligned to 1F0Y. Structure of BCKDHA is shown in green.






Discussion
Evaluation of alignments using structures
Results
The following table shows an overview of the structures used for building models, the scores of the structural alignment (RMSD and LGA_S - structure similarity score, both according to the Cα atoms), and the scores of the sequence alignment (E-value, probability and sequence identity).
| model | RMSD | LGA_S | E-value | probability | sequence identity |
|---|---|---|---|---|---|
| 1qs0 | 1.24 | 84.085 | 5.8E-94 | 100.0 | 38 |
| 1w85 | 1.77 | 78.356 | 8.3E-87 | 100.0 | 33 |
| 2ozl | 1.63 | 74.027 | 3.2E-69 | 100.0 | 27 |
| 2yic | 2.45 | 41.732 | 5.7E-47 | 100.0 | 16 |
| 3l84 | 2.01 | 32.412 | 6.5E-18 | 99.5 | 21 |
| 2q28 | 1.86 | 25.398 | 1.6E-08 | 97.9 | 13 |
| 1r9j | 1.73 | 30.095 | 1.1E-06 | 97.2 | 25 |
| 2vk8 | 2.12 | 21.990 | 3.7E-05 | 96.4 | 22 |
| 1t9b | 1.83 | 23.724 | 0.0011 | 94.9 | 18 |
| 2c31 | 2.00 | 21.849 | 0.011 | 92.7 | 21 |
| E-value | log10(E-value) | probability | sequence identity | |
|---|---|---|---|---|
| RMSD | 0.15 | 0.49 | -0.19 | -0.74 |
| LGA_S | -0.33 | -0.98 | 0.71 | 0.82 |
As can be seen in the above table, the RMSD has a weak correlation to the logarithm of E-value and a higher correlation to sequence identity. The RMSD is lower, if the E-value is lower or the sequence identity is higher.
The same tendency can be seen for the LGA_S score, but here the correlations are higher. The LGA_S score shows also a correlation to the probability in contrast to the RMSD.
The signs are opposite for RMSD and LGA_S, because the RMSD is lower for higher similarity, but the LGA_S is higher.
The relationship of LGA_S and E-value, the pair of scores with the highest correlation, for the 10 models is shown in the following plot.

Discussion
The correlations between structural and sequence alignment scores are as expected. A low E-value indicates a hit that is unlikely to occur only by chance, so it is significant. This means it is related to the query and will have a similar structure. So the RMSD, which measures the difference (root mean squared distance) in the aligned structures, will be low for nearly related proteins. Also if two sequences have a high sequence identity, they will more likely have the same structure, which explains the correlation of RMSD to it. The reason for the observed correlations of RMSD to the alignment scores being weaker than those of the LGA_S score, could be that the RMSD is calculated only locally for structurally aligned residues. So it tends to be too low, because a protein pair which has a very similar part but another dissimilar, not alignable part, would have a low RMSD. For probability the values in the sample don't cover the whole range of possible values, so there was observed almost no correlation to RMSD. We did not take very distant relatives to create structure models, if they had a too high E-value.
The LGA_S score, which combines local and global distances to the reference structure (here: the structure of our BCKDHA protein), gives a better indication of the overall structure similarity, than the local RMSD. It is correlated to the sequence alignment scores (in particular to the logarithm of E-value), so a significant hit in the sequence alignment is likely to have a structure similar to those of the query, and thus can be used to create a model of the structure of the query protein.
All observed local RMSD values vere relatively low and also the LGA_S values were high at least for nearly related structures. So the approach of simply copying Cα atom coordinates of aligned residues from the structure of a related sequence helps to build a model of the unknown structure of a protein.