Difference between revisions of "Fabry:Sequence-based analyses"
Rackersederj (talk | contribs) (→Signal peptides: Added it all. YEAH) |
Rackersederj (talk | contribs) (→GO terms: This is it. Weekend here I come!) |
||
| Line 749: | Line 749: | ||
== GO terms == |
== GO terms == |
||
| + | === QuickGO === |
||
| + | |||
| + | <div style="float:right; border:thin solid lightgrey; margin: 20px;"> |
||
| + | <figtable id="tab:QuickGO"> |
||
| + | <caption>Results of the QuickGO search</caption> |
||
| + | {| style="border-collapse: collapse; border-width: 1px; border-style: solid; border-color: #000" |
||
| + | ! style="border-style: solid; border-width: 1px;"|Code |
||
| + | ! style="border-style: solid; border-width: 1px;"|Name |
||
| + | |- |
||
| + | | style="border-style: solid; border-width: 1px"| GO:0052692 |
||
| + | | style="border-style: solid; border-width: 1px"| raffinose alpha-galactosidase activity |
||
| + | |- |
||
| + | | style="border-style: solid; border-width: 1px"| GO:0051001 |
||
| + | | style="border-style: solid; border-width: 1px"| negative regulation of nitric-oxide synthase activity |
||
| + | |- |
||
| + | | style="border-style: solid; border-width: 1px"| GO:0046479 |
||
| + | | style="border-style: solid; border-width: 1px"| glycosphingolipid catabolic process |
||
| + | |- |
||
| + | | style="border-style: solid; border-width: 1px"| GO:0046477 |
||
| + | | style="border-style: solid; border-width: 1px"| glycosylceramide catabolic process |
||
| + | |- |
||
| + | | style="border-style: solid; border-width: 1px"| GO:0045019 |
||
| + | | style="border-style: solid; border-width: 1px"| negative regulation of nitric oxide biosynthetic process |
||
| + | |- |
||
| + | | style="border-style: solid; border-width: 1px"| GO:0044281 |
||
| + | | style="border-style: solid; border-width: 1px"| small molecule metabolic process |
||
| + | |- |
||
| + | | style="border-style: solid; border-width: 1px"| GO:0043202 |
||
| + | | style="border-style: solid; border-width: 1px"| lysosomal lumen |
||
| + | |- |
||
| + | | style="border-style: solid; border-width: 1px"| GO:0043169 |
||
| + | | style="border-style: solid; border-width: 1px"| cation binding |
||
| + | |- |
||
| + | | style="border-style: solid; border-width: 1px"| GO:0042803 |
||
| + | | style="border-style: solid; border-width: 1px"| protein homodimerization activity |
||
| + | |- |
||
| + | | style="border-style: solid; border-width: 1px"| GO:0016936 |
||
| + | | style="border-style: solid; border-width: 1px"| galactoside binding |
||
| + | |- |
||
| + | | style="border-style: solid; border-width: 1px"| GO:0016798 |
||
| + | | style="border-style: solid; border-width: 1px"| hydrolase activity, acting on glycosyl bonds |
||
| + | |- |
||
| + | | style="border-style: solid; border-width: 1px"| GO:0016787 |
||
| + | | style="border-style: solid; border-width: 1px"| hydrolase activity |
||
| + | |- |
||
| + | | style="border-style: solid; border-width: 1px"| GO:0016139 |
||
| + | | style="border-style: solid; border-width: 1px"| glycoside catabolic process |
||
| + | |- |
||
| + | | style="border-style: solid; border-width: 1px"| GO:0009311 |
||
| + | | style="border-style: solid; border-width: 1px"| oligosaccharide metabolic process |
||
| + | |- |
||
| + | | style="border-style: solid; border-width: 1px"| GO:0008152 |
||
| + | | style="border-style: solid; border-width: 1px"| metabolic process |
||
| + | |- |
||
| + | | style="border-style: solid; border-width: 1px"| GO:0006687 |
||
| + | | style="border-style: solid; border-width: 1px"| glycosphingolipid metabolic process |
||
| + | |- |
||
| + | | style="border-style: solid; border-width: 1px"| GO:0006665 |
||
| + | | style="border-style: solid; border-width: 1px"| sphingolipid metabolic process |
||
| + | |- |
||
| + | | style="border-style: solid; border-width: 1px"| GO:0005975 |
||
| + | | style="border-style: solid; border-width: 1px"| carbohydrate metabolic process |
||
| + | |- |
||
| + | | style="border-style: solid; border-width: 1px"| GO:0005794 |
||
| + | | style="border-style: solid; border-width: 1px"| Golgi apparatus |
||
| + | |- |
||
| + | | style="border-style: solid; border-width: 1px"| GO:0005764 |
||
| + | | style="border-style: solid; border-width: 1px"| lysosome |
||
| + | |- |
||
| + | | style="border-style: solid; border-width: 1px"| GO:0005737 |
||
| + | | style="border-style: solid; border-width: 1px"| cytoplasm |
||
| + | |- |
||
| + | | style="border-style: solid; border-width: 1px"| GO:0005625 |
||
| + | | style="border-style: solid; border-width: 1px"| soluble fraction |
||
| + | |- |
||
| + | | style="border-style: solid; border-width: 1px"| GO:0005576 |
||
| + | | style="border-style: solid; border-width: 1px"| extracellular region |
||
| + | |- |
||
| + | | style="border-style: solid; border-width: 1px"| GO:0005515 |
||
| + | | style="border-style: solid; border-width: 1px"| protein binding |
||
| + | |- |
||
| + | | style="border-style: solid; border-width: 1px"| GO:0005102 |
||
| + | | style="border-style: solid; border-width: 1px"| receptor binding |
||
| + | |- |
||
| + | | style="border-style: solid; border-width: 1px"| GO:0004557 |
||
| + | | style="border-style: solid; border-width: 1px"| alpha-galactosidase activity |
||
| + | |- |
||
| + | | style="border-style: solid; border-width: 1px"| GO:0004553 |
||
| + | | style="border-style: solid; border-width: 1px"| hydrolase activity, hydrolyzing O-glycosyl compounds |
||
| + | |- |
||
| + | | style="border-style: solid; border-width: 1px"| GO:0003824 |
||
| + | | style="border-style: solid; border-width: 1px"| catalytic activity |
||
| + | |- |
||
| + | |} |
||
| + | </figtable> |
||
| + | </div> |
||
| + | Since we used [http://www.ebi.ac.uk/QuickGO QuickGO] in [[Fabry:Sequence_alignments_(sequence_searches_and_multiple_alignments) | Task2]] to download the GO terms we decided to refine our [[Fabry:Sequence_alignments_(sequence_searches_and_multiple_alignments)#Sequence_searches | GO analysis]] now. The QuickGO search revealed 28 distinct GO terms (see <xr id="tab:QuickGO"/>). A lot of these are about hydrolase activity and metabolic processes (especially [[Media:Fabry-glycosphingolipid_biosynthesis_globoseries.png | glycosphingolipid biosynthesis]]), as well as the location of the protein (lysosome) and its enzymatic function. The rather large number of resulting GO terms, may be explained by the listing of ''all'' possible terms, especially the quite general ones. |
||
| + | |||
| + | <br style="clear:both;"> |
||
=== GOPET === |
=== GOPET === |
||
| − | Searching the GOPET annotation tool with the AGAL_HUMAN [[Alpha-galactosidase_sequence|sequence]] revealed 5 GOIds, which are displayed in <xr id="tab:GOPET"/>. On a first glance, since we already know the name and function of the protein, it is a bit surprising, that alpha-galactosidase activity is only the third entry with 96% confidence. In our already carried out information gathering we learned that α-galactosidase A is a hydrolase thus the first three entries were not surprising. Considering that our enzyme mainly is a glycosidase, the both entries on top of the list make perfekt sense.<br> |
||
| − | Again a bit surprising was the last entry. α-N-Acetylgalactosaminidase is actually used for enzyme replacement therapy, which we mention on our [[Fabry_Disease | main page]]. The structure of both enzymes is similar to each other, but this still does not explain the association of this GO term to the AGAL protein. |
||
| + | <div style="float:right; border:thin solid lightgrey; margin: 20px;"> |
||
<figtable id="tab:GOPET"> |
<figtable id="tab:GOPET"> |
||
| − | <caption> |
+ | <caption>Results of the GOPET search</caption> |
{| style="border-collapse: collapse; border-width: 1px; border-style: solid; border-color: #000" |
{| style="border-collapse: collapse; border-width: 1px; border-style: solid; border-color: #000" |
||
! style="border-style: solid; border-width: 1px;" colspan="6"|Result for GOPET search |
! style="border-style: solid; border-width: 1px;" colspan="6"|Result for GOPET search |
||
| + | |- |
||
| − | |- align="center" |
||
! style="border-style: solid; border-width: 1px"| GOid |
! style="border-style: solid; border-width: 1px"| GOid |
||
! style="border-style: solid; border-width: 1px"| Aspect |
! style="border-style: solid; border-width: 1px"| Aspect |
||
! style="border-style: solid; border-width: 1px"| Confidence |
! style="border-style: solid; border-width: 1px"| Confidence |
||
! style="border-style: solid; border-width: 1px"| GO term |
! style="border-style: solid; border-width: 1px"| GO term |
||
| + | |- |
||
| − | |- align="center" |
||
| style="border-style: solid; border-width: 1px"| [http://www.dkfz.de/menu/cgi-bin/srs/wgetz?-newId+-e+%5bGO-ID:0016798%5d GO:0016798] |
| style="border-style: solid; border-width: 1px"| [http://www.dkfz.de/menu/cgi-bin/srs/wgetz?-newId+-e+%5bGO-ID:0016798%5d GO:0016798] |
||
| style="border-style: solid; border-width: 1px"| Molecular Function Ontology (F) |
| style="border-style: solid; border-width: 1px"| Molecular Function Ontology (F) |
||
| − | | style="border-style: solid; border-width: 1px"| 98% |
+ | | style="border-style: solid; border-width: 1px; text-align: right"| 98% |
| style="border-style: solid; border-width: 1px"| hydrolase activity acting on glycosyl bonds |
| style="border-style: solid; border-width: 1px"| hydrolase activity acting on glycosyl bonds |
||
| + | |- |
||
| − | |- align="center" |
||
| style="border-style: solid; border-width: 1px"| [http://www.dkfz.de/menu/cgi-bin/srs/wgetz?-newId+-e+%5bGO-ID:0004553%5d GO:0004553] |
| style="border-style: solid; border-width: 1px"| [http://www.dkfz.de/menu/cgi-bin/srs/wgetz?-newId+-e+%5bGO-ID:0004553%5d GO:0004553] |
||
| style="border-style: solid; border-width: 1px"| Molecular Function Ontology (F) |
| style="border-style: solid; border-width: 1px"| Molecular Function Ontology (F) |
||
| − | | style="border-style: solid; border-width: 1px"| 98% |
+ | | style="border-style: solid; border-width: 1px; text-align: right"| 98% |
| style="border-style: solid; border-width: 1px"| hydrolase activity hydrolyzing O-glycosyl compounds |
| style="border-style: solid; border-width: 1px"| hydrolase activity hydrolyzing O-glycosyl compounds |
||
| + | |- |
||
| − | |- align="center" |
||
| style="border-style: solid; border-width: 1px"| [http://www.dkfz.de/menu/cgi-bin/srs/wgetz?-newId+-e+%5bGO-ID:0016787%5d GO:0016787] |
| style="border-style: solid; border-width: 1px"| [http://www.dkfz.de/menu/cgi-bin/srs/wgetz?-newId+-e+%5bGO-ID:0016787%5d GO:0016787] |
||
| style="border-style: solid; border-width: 1px"| Molecular Function Ontology (F) |
| style="border-style: solid; border-width: 1px"| Molecular Function Ontology (F) |
||
| − | | style="border-style: solid; border-width: 1px"| 97% |
+ | | style="border-style: solid; border-width: 1px; text-align: right"| 97% |
| style="border-style: solid; border-width: 1px"| hydrolase activity |
| style="border-style: solid; border-width: 1px"| hydrolase activity |
||
| + | |- |
||
| − | |- align="center" |
||
| style="border-style: solid; border-width: 1px"| [http://www.dkfz.de/menu/cgi-bin/srs/wgetz?-newId+-e+%5bGO-ID:0004557%5d GO:0004557] |
| style="border-style: solid; border-width: 1px"| [http://www.dkfz.de/menu/cgi-bin/srs/wgetz?-newId+-e+%5bGO-ID:0004557%5d GO:0004557] |
||
| style="border-style: solid; border-width: 1px"| Molecular Function Ontology (F) |
| style="border-style: solid; border-width: 1px"| Molecular Function Ontology (F) |
||
| − | | style="border-style: solid; border-width: 1px"| 96% |
+ | | style="border-style: solid; border-width: 1px; text-align: right"| 96% |
| style="border-style: solid; border-width: 1px"| alpha-galactosidase activity |
| style="border-style: solid; border-width: 1px"| alpha-galactosidase activity |
||
| + | |- |
||
| − | |- align="center" |
||
| style="border-style: solid; border-width: 1px"| [http://www.dkfz.de/menu/cgi-bin/srs/wgetz?-newId+-e+%5bGO-ID:0008456%5d GO:0008456] |
| style="border-style: solid; border-width: 1px"| [http://www.dkfz.de/menu/cgi-bin/srs/wgetz?-newId+-e+%5bGO-ID:0008456%5d GO:0008456] |
||
| style="border-style: solid; border-width: 1px"| Molecular Function Ontology (F) |
| style="border-style: solid; border-width: 1px"| Molecular Function Ontology (F) |
||
| − | | style="border-style: solid; border-width: 1px"| 89% |
+ | | style="border-style: solid; border-width: 1px; text-align: right"| 89% |
| style="border-style: solid; border-width: 1px"| alpha-N-acetylgalactosaminidase activity |
| style="border-style: solid; border-width: 1px"| alpha-N-acetylgalactosaminidase activity |
||
|- |
|- |
||
|} |
|} |
||
</figtable> |
</figtable> |
||
| + | </div> |
||
| + | Searching the GOPET annotation tool with the AGAL_HUMAN [[Alpha-galactosidase_sequence|sequence]] revealed 5 GOIds, which are displayed in <xr id="tab:GOPET"/>. Even maximizing the "maximum number of GO prediction to be displayed per sequence" and minimizing the "confidence threshold for prediction" did not result in any more sequences.<br> |
||
| − | === ProtFun2.0 === |
||
| + | On a first glance, since we already know the name and function of the protein, it is a bit surprising, that alpha-galactosidase activity is only the third entry with 96% confidence. In our already carried out information gathering we learned that α-galactosidase A is a hydrolase thus the first three entries were not surprising. Considering that our enzyme mainly is a glycosidase, the both entries on top of the list make perfekt sense.<br> |
||
| − | EC=3.2.1.22(EC 3.-.-.- Hydrolase)<br> |
||
| + | Again a bit surprising was the last entry. α-N-Acetylgalactosaminidase is actually used for enzyme replacement therapy, which we mention on our [[Fabry_Disease | main page]]. The structure of both enzymes is similar to each other, but this still does not explain the association of this GO term to the AGAL protein. Also, the [[Fabry:Sequence-based_analyses#QuickGO |QuickGO]] search did not bring up this GO term.<br> |
||
| − | Predicted: EC 6.-.-.-(Ligase) |
||
| − | ############## ProtFun 2.2 predictions ############## |
||
| − | |||
| − | >gi_4504009_ |
||
| − | |||
| − | # Functional category Prob Odds |
||
| − | Amino_acid_biosynthesis 0.283 12.847 |
||
| − | Biosynthesis_of_cofactors 0.339 4.708 |
||
| − | Cell_envelope => 0.652 10.690 |
||
| − | Cellular_processes 0.057 0.783 |
||
| − | Central_intermediary_metabolism 0.400 6.343 |
||
| − | Energy_metabolism 0.151 1.678 |
||
| − | Fatty_acid_metabolism 0.032 2.448 |
||
| − | Purines_and_pyrimidines 0.506 2.082 |
||
| − | Regulatory_functions 0.013 0.083 |
||
| − | Replication_and_transcription 0.047 0.175 |
||
| − | Translation 0.211 4.807 |
||
| − | Transport_and_binding 0.549 1.339 |
||
| − | |||
| − | # Enzyme/nonenzyme Prob Odds |
||
| − | Enzyme => 0.805 2.811 |
||
| − | Nonenzyme 0.195 0.273 |
||
| − | |||
| − | # Enzyme class Prob Odds |
||
| − | Oxidoreductase (EC 1.-.-.-) 0.176 0.845 |
||
| − | Transferase (EC 2.-.-.-) 0.195 0.564 |
||
| − | Hydrolase (EC 3.-.-.-) 0.244 0.769 |
||
| − | Lyase (EC 4.-.-.-) 0.029 0.608 |
||
| − | Isomerase (EC 5.-.-.-) 0.010 0.321 |
||
| − | Ligase (EC 6.-.-.-) => 0.141 2.776 |
||
| − | |||
| − | # Gene Ontology category Prob Odds |
||
| − | Signal_transducer 0.090 0.419 |
||
| − | Receptor 0.014 0.083 |
||
| − | Hormone 0.002 0.318 |
||
| − | Structural_protein 0.004 0.127 |
||
| − | Transporter 0.024 0.222 |
||
| − | Ion_channel 0.010 0.169 |
||
| − | Voltage-gated_ion_channel 0.003 0.127 |
||
| − | Cation_channel 0.010 0.215 |
||
| − | Transcription 0.047 0.367 |
||
| − | Transcription_regulation 0.026 0.204 |
||
| − | Stress_response 0.049 0.552 |
||
| − | Immune_response 0.012 0.136 |
||
| − | Growth_factor 0.006 0.412 |
||
| − | Metal_ion_transport 0.009 0.020 |
||
| − | |||
| − | // |
||
| + | <br style="clear:both;"> |
||
| + | === ProtFun2.2 === |
||
| + | In the [https://www.dropbox.com/s/sn9n8h4gojhxvkn/ProtFun_pred.txt output file of ProtFun], the "=>" indicates, which of the subcategories of each category is predicted to be true for the submitted sequence. This prediction is performed on basis of the highest information content. In the pictures below the probabilty, odds and [http://en.wikipedia.org/wiki/Self-information information content] (probabilty*odds) are shown separately for each category. The left y-axis is assigned to the probabilty (blue) and the information content (red line), the right y-axis is assigned to the probabilty, which is multiplied by 10 for a better perceptibility. <br> |
||
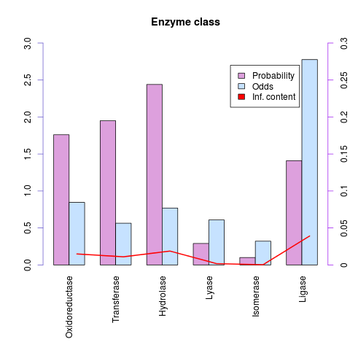
| + | According to the information content the most likely functional category of the human α-galactosidase A protein is "Cell envelope". Considering our own researches, we would not come to this conclusion, but rather assign a metabolic or regulatory class, since to us known GO terms are for example "glycoside catabolic process", "negative regulation of nitric oxide biosynthetic process" and "small molecule metabolic process".<br> |
||
| + | The prediction that the submitted sequence belongs to an enzyme is right. <br> |
||
| + | In our opinion, it is not always the best evaluation method to decide on the basis of the information content, since the chosen enzyme class "Ligase" indeed has the highest information content, but not the highest probabilty and is, considering literature, wrong. AGAL_HUMAN clearly is a Hydrolase, which is also indicated by the high probabilty. The assigned EC number of the galactosidase is EC=3.2.1.22.<br> |
||
| + | ProtFun does not predict a Gene Ontology category, since the category with the highest information content has odds lower than 1. We actually expected this category to be very unclear since the protein has very diverse cellular functions. |
||
| + | |||
| + | <gallery widths=350px heights=350px perrow=3 caption="Probability, Odds and Information Content of the ProtFun2.2 predictions"> |
||
| + | File:FABRY_Functional_category-barplot.png | Functional category |
||
| + | File:FABRY_Enzyme_class-barplot.png | Enzyme class |
||
| + | File:FABRY_Gene_Ontology_category-barplot.png | Gene Ontology category |
||
| + | </gallery> |
||
| + | |||
| + | ProtFun uses a lot of other ressources to predict the probabilties and odds of the categories. Some of the outcomes are listed in <xr id="tab:ProtFun"/>. It is known that the α-galactosidase A protein has a 31 residues long signal peptide on position 1 to 31 which later is cleaved of. Thus the prediction of SignalP 3.0 is right.<br> |
||
| + | The prediction of a propeptide cleavage site could not be confirmed in the human AGAL protein.<br> |
||
| + | ProtFun infered 22 phosphorylation sites at the positions 62, 102, 201, 235, 238, 241, 276, 304, 364, 371, 405, 424, 366, 400, 86, 134, 151, 152, 173, 184, 207, 216. [http://www.phosphosite.org/proteinAction.do?id=12871&showAllSites=true Phosphosite] confirms phosphorylation modificatopns at S23, Y134 and H186 of which only one was predicted by NetPhos.<br> |
||
| + | [http://www.uniprot.org/uniprot/P06280 Uniprot] claims that at the positions 139, 192, 215 and 408 there are N-linked Glycosilated sites (the last one is only a potential site). All 4 are predicted by NetNGlyc. As predicted by NetOGlyc, there are no (known) O-glycosylated sites.<br> |
||
| + | See also sections [[Fabry:Sequence-based_analyses#Transmembrane_helices | Transmembrane helices]] and [[Fabry:Sequence-based_analyses#Signal_peptides| Signal peptides]] for further information on these two topics. |
||
| + | |||
| + | <div style="float:right; border:thin solid lightgrey; margin: 20px;"> |
||
| + | <figtable id="tab:ProtFun"> |
||
| + | <caption>Output rendered by the individual features used by ProtFun 2.2</caption> |
||
| + | {| style="border-collapse: collapse; border-width: 1px; border-style: solid; border-color: #000" |
||
| + | ! style="border-style: solid; border-width: 1px"| Feature |
||
| + | ! style="border-style: solid; border-width: 1px"| Output summary |
||
| + | ! style="border-style: solid; border-width: 1px"| Details |
||
| + | |- |
||
| + | | style="border-style: solid; border-width: 1px"| SignalP 3.0 |
||
| + | | style="border-style: solid; border-width: 1px"| Most likely cleavage site between pos. 31 and 32: ARA-LD |
||
| + | | style="border-style: solid; border-width: 1px"| Using neural networks (NN) and hidden Markov models (HMM) trained on eukaryotes |
||
| + | |- |
||
| + | | style="border-style: solid; border-width: 1px"| ProP 1.0 |
||
| + | | style="border-style: solid; border-width: 1px"| 1 propeptide cleavage site predicted at position: 196 |
||
| + | | style="border-style: solid; border-width: 1px"| Furin-type cleavage site prediction (Arginine/Lysine residues) |
||
| + | |- |
||
| + | | style="border-style: solid; border-width: 1px"| TargetP 1.1 |
||
| + | | style="border-style: solid; border-width: 1px"| No high confidence targeting predition |
||
| + | | style="border-style: solid; border-width: 1px"| - |
||
| + | |- |
||
| + | | style="border-style: solid; border-width: 1px"| NetPhos 2.0 |
||
| + | | style="border-style: solid; border-width: 1px"| 22 putative phosphorylation sites |
||
| + | | style="border-style: solid; border-width: 1px"| phosphorylation site prediction |
||
| + | |- |
||
| + | | style="border-style: solid; border-width: 1px"| NetOGlyc 3.1 |
||
| + | | style="border-style: solid; border-width: 1px"| No O-glycosylated sites predicted |
||
| + | | style="border-style: solid; border-width: 1px"| - |
||
| + | |- |
||
| + | | style="border-style: solid; border-width: 1px"| NetNGlyc 1.0 |
||
| + | | style="border-style: solid; border-width: 1px"| 4 putative N-glycosylated sites at positions 139 192 215 408 |
||
| + | | style="border-style: solid; border-width: 1px"| - |
||
| + | |- |
||
| + | | style="border-style: solid; border-width: 1px"| TMHMM 2.0 |
||
| + | | style="border-style: solid; border-width: 1px"| No TM helices predicted |
||
| + | | style="border-style: solid; border-width: 1px"| - |
||
| + | |- |
||
| + | |} |
||
| + | </figtable> |
||
| + | </div> |
||
| + | |||
| + | <br style="clear:both;"> |
||
=== Pfam === |
=== Pfam === |
||
| + | |||
| + | The Pfam sequence search revealed one significant Pfam-A match, which is shown in <xr id="fig:pfam_fam"/>. |
||
| + | |||
| + | <div style="float:right; border:thin solid lightgrey; margin: 20px;"> |
||
| + | <figtable id="fig:pfam_fam"> |
||
| + | <caption>Pfam-A match</caption> |
||
| + | {| style="border-collapse: collapse; border-width: 1px; border-style: solid; border-color: #000" |
||
| + | ! style="border-style: solid; border-width: 1px"| Family |
||
| + | ! style="border-style: solid; border-width: 1px"| Description |
||
| + | ! style="border-style: solid; border-width: 1px"| Entry xtype |
||
| + | ! style="border-style: solid; border-width: 1px"| Clan |
||
| + | ! style="border-style: solid; border-width: 1px"| Envelope Start |
||
| + | ! style="border-style: solid; border-width: 1px"| Envelope End |
||
| + | ! style="border-style: solid; border-width: 1px"| Alignment Start |
||
| + | ! style="border-style: solid; border-width: 1px"| Alignment End |
||
| + | ! style="border-style: solid; border-width: 1px"| HMM From |
||
| + | ! style="border-style: solid; border-width: 1px"| HMM To |
||
| + | ! style="border-style: solid; border-width: 1px"| Bit score |
||
| + | ! style="border-style: solid; border-width: 1px"| E-value |
||
| + | |- |
||
| + | | style="border-style: solid; border-width: 1px"| Melibiase |
||
| + | | style="border-style: solid; border-width: 1px"| Melibiase |
||
| + | | style="border-style: solid; border-width: 1px"| Family |
||
| + | | style="border-style: solid; border-width: 1px"| CL0058 |
||
| + | | style="border-style: solid; border-width: 1px"| 33 |
||
| + | | style="border-style: solid; border-width: 1px"| 149 |
||
| + | | style="border-style: solid; border-width: 1px"| 40 |
||
| + | | style="border-style: solid; border-width: 1px"| 146 |
||
| + | | style="border-style: solid; border-width: 1px"| 41 |
||
| + | | style="border-style: solid; border-width: 1px"| 140 |
||
| + | | style="border-style: solid; border-width: 1px"| 50.0 |
||
| + | | style="border-style: solid; border-width: 1px"| 1.5e-13 |
||
| + | |- |
||
| + | |} |
||
| + | </figtable> |
||
| + | </div> |
||
| + | |||
| + | <br style="clear:both;"> |
||
| + | <figure id="fig:AGAL">[[File:Alpha_GAL_A.png|200px|thumb|right|<caption>The protein encoded by the Fabry-associated GLA gene: [[Alpha-galactosidase|α-Galactosidase A]]</caption>]]</figure> |
||
| + | |||
| + | |||
| + | |||
| + | The alpha-galactosidase A protein is, according to PFAM, in the family of the [http://pfam.sanger.ac.uk/family/PF02065 Melibiases] (see <xr id="tab:Pfam_Melibiose"/>). |
||
| + | The Pfam PF02065 family itself is refered to as "Glycoside hydrolase family 27" and "Glycoside hydrolase family 36", the AGAL_HUMAN falls into the first category. Their common characteristic is that the members of this family are glycoside hydrolases (EC 3.2.1.). The AGAL enzyme catalyzes the hydrolisis of the disaccharide Melibiose (D-Gal-α(1→6)-D-Glc) into its two components galactose and glucose.<br> |
||
| + | <div style="float:left; border:thin solid lightgrey; margin: 20px;"> |
||
| + | <figtable id="tab:Pfam_Melibiose"> |
||
| + | <caption>Melibiase Identifiers</caption> |
||
| + | {| style="border-collapse: collapse; border-width: 1px; border-style: solid; border-color: #000" |
||
| + | ! style="border-style: solid; border-width: 1px;" colspan="6"|Melibiase Identifiers |
||
| + | |- |
||
| + | ! style="border-style: solid; border-width: 1px"| Symbol |
||
| + | | style="border-style: solid; border-width: 1px"| Melibiase |
||
| + | |- |
||
| + | ! style="border-style: solid; border-width: 1px"| Pfam |
||
| + | | style="border-style: solid; border-width: 1px"| [http://pfam.sanger.ac.uk/family?acc=PF02065 PF02065] |
||
| + | |- |
||
| + | ! style="border-style: solid; border-width: 1px"| Pfam clan |
||
| + | | style="border-style: solid; border-width: 1px"| [http://pfam.sanger.ac.uk/clan/CL0058 CL0058] |
||
| + | |- |
||
| + | ! style="border-style: solid; border-width: 1px"| InterPro |
||
| + | | style="border-style: solid; border-width: 1px"| [http://www.ebi.ac.uk/interpro/DisplayIproEntry?ac=IPR000111 IPR000111] |
||
| + | |- |
||
| + | ! style="border-style: solid; border-width: 1px"| SCOP |
||
| + | | style="border-style: solid; border-width: 1px"| [http://scop.mrc-lmb.cam.ac.uk/scop/search.cgi?tlev=fa;&pdb=1ktc 1ktc] |
||
| + | |- |
||
| + | ! style="border-style: solid; border-width: 1px"| SUPERFAMILY |
||
| + | | style="border-style: solid; border-width: 1px"| [http://supfam.org/SUPERFAMILY/cgi-bin/search.cgi?search_field=1ktc 1ktc] |
||
| + | |- |
||
| + | ! style="border-style: solid; border-width: 1px"| CAZy |
||
| + | | style="border-style: solid; border-width: 1px"| [http://www.cazy.org/GH27.html GH27] |
||
| + | |- |
||
| + | |} |
||
| + | </figtable> |
||
| + | </div> |
||
| + | |||
| + | The alignment of positions 40 - 146 of the AGAL_HUMAN protein sequence and the matching HMM used in this prediction, is shown in <xr id="fig:pfam_ali" />. According to the color code, indicating the degree of confidence of each aligned position, there is an overall very good agreement of the Hidden Markov Model and the sequence, except for the residues 17-46. Checking our background knowledge and the [http://www.uniprot.org/uniprot/P06280 Uniprot database] we could not find a very interesting or abnormal region here, but the signal peptide cleavage site and a beta strand at position 42-46.<br> |
||
| + | Our query protein belongs to a rather large Clan, the Glyco_hydro_tim Clan ([http://pfam.sanger.ac.uk/clan/CL0058 CL0058]), which includes 4 CAZy-Clans (GH-A, GH-D, GH-H and GH-K). They main attribute of all the included glycosyl hydrolase enzymes is the hold of a a TIM barrel fold (eight α-helices and eight parallel β-strands that alternate along the peptide backbone [http://en.wikipedia.org/wiki/TIM_barrel source]). This fold (residue 31 - 324) can be well seen in <xr id="fig:AGAL"/>, where the 3D structure of α-Galactosidase A is depicted.<br> |
||
| + | The InterPro protein sequence analysis and classification assigns the enzyme to the "IPR000111 Glycoside hydrolase, clan GH-D" family. As mentioned before, GH-D is part of the Glyco_hydro_tim Clan and thus it is not surprising, that the description is almost the same. According to InterPro, there are 6 IPR000111 family members in the human body.<br> |
||
| + | The Structural Classification of Proteins (SCOP) finds two domains, an Amylase, catalytic domain ([http://scop.mrc-lmb.cam.ac.uk/scop/data/scop.b.d.b.j.b.html c.1.8.1], residues 32 - 323) and an alpha-Amylases, C-terminal beta-sheet domain ([http://scop.mrc-lmb.cam.ac.uk/scop/data/scop.b.c.bcb.b.b.html b.71.1.1], residues 324 - 421). The catalytic domain is an alpha and beta protein (a/b) with TIM beta/alpha-barrel fold, which is consonant with the affiliation to the Glyco_hydro_tim Clan. The C-terminal beta-sheet domain is an all beta protein, the fold and superfamily is Glycosyl hydrolase domain. This is also not surprising to us.<br> |
||
| + | AGAL is in the CAZy GH27 ([http://www.cazy.org/GH27.html Glycoside Hydrolase Family 27]) family, together with a-N-acetylgalactosaminidase (EC 3.2.1.49; as mentioned before, used for enzyme replacement therapy), isomalto-dextranase(EC 3.2.1.94) and b-L-arabinopyranosidase (EC 3.2.1.88). See http://www.cazypedia.org/index.php/Glycoside_Hydrolase_Family_27 TODO |
||
| + | |||
| + | <figure id="fig:pfam_ali">[[File:pfam_alignment.png|772px|left|thumb|<caption>Melibiase - alignment region residues 40 - 146</caption>]]</figure> |
||
| + | |||
| + | <br style="clear:both;"> |
||
== Other programs and ressources == |
== Other programs and ressources == |
||
Revision as of 20:04, 18 May 2012
Fabry Disease » Sequence-based analyses
The following analyses were performed on the basis of the α-Galactosidase A sequence. Please consult the journal for the commands used to generate the results.
Contents
Secondary structure
Disorder
Transmembrane helices
α-Galactosidase A (P06280)
<figure id="fig:1r46_TM">

</figure> <figure id="fig:1r46_TMHMM">

</figure>
| organism: | Homo sapiens (Human) |
|---|---|
| pdb-id: | 1r46 |
Since the whole sequence is labeled as "NON CYTOPLASMIC", the prediction of Polyphobius is that it contains no membrane helices (see Polyphobius output file). This result is consistent with both databases, OPM and PDBTM and the TMHMM-2.0 prediction (see <xr id="fig:1r46_TMHMM" />).
In <xr id="fig:1r46_TM"/> the posterior probabilities of cytoplasmic(green), non cytoplasmic(blue), TM helix(grey area) and signal peptide(red) are shown. The grey area indicates weak evidence for a transmembrane helix which where not predicted. The first small peak can be observed in both pictures. Considering the shown probabilities and our background knowledge, the prediction seems to be true. Only the end of the signal peptide at residue 29 is, according to our knowledge, too early and should be shifted to position 31.
D(3) dopamine receptor (P35462)
<figtable id="tab:3pbl_TM_PostProb"> Posterior probability plots Polyphobius and TMHMM
 Human D(3) dopamine receptor, plot of posterior probabilities of features predicted by Polyphobius |
 Human D(3) dopamine receptor, plot of posterior probabilities of features predicted by TMHMM |
</figtable>
| organism: | Homo sapiens (Human) |
|---|---|
| pdb-id: | 3pbl (only available structure) |
Polyphobius predicts 7 transmembrane regions. Comparing this result to the pictures in <xr id="tab:3pbl_TM"/>, we can again see a consistent result with the two databases. The only difference between the models seems to be the number of residues on the cytosolic side of the membrane (green in the right picture) of this hydrolase/hydrolase inhibitor. Inquiring the PDBTM site, this area is marked as residues 1002-1161, which do not even belong to the 400 aa long protein. This might result from the experiment, the structure was derived from. It says, that the protein was enginered (see source1 and source2)
Besides from that, a difference in predicted and observed starting and end points could be revealed (see <xr id="tab:3pbl_start_end"/>), mainly those of PDBTM, which leads to a great difference in its mean length compared to the other methods (see <xr id="tab:3pbl_TM"/>, picture 3). Here, PDBTM throughout assigns shorter helices, since its helices always start later and end earlier, which can also be seen in <xr id="fig:bitshift" />. The boundaries Polyphobius predicted may be seen in the Polyphobius output file.
The posterior probability of the first 6 predicted helices is very similar, as one can see in <xr id="tab:3pbl_TM_PostProb"/>. The only difference is in the last TM, where the probability assigned by TMHMM is nearly 90%, whereas Polyphobius only assigns about 55%.
<figtable id="tab:3pbl_start_end"> Start and end points of each transmembrane helix predicted
| TMH number | 1 | 2 | 3 | 4 | 5 | 6 | 7 | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Method | Length | Start | End | Length | Start | End | Length | Start | End | Length | Start | End | Length | Start | End | Length | Start | End | Length | Start | End |
| Polyphobius | 26 | 30 | 55 | 23 | 66 | 88 | 22 | 105 | 126 | 21 | 150 | 170 | 25 | 188 | 212 | 24 | 329 | 352 | 20 | 367 | 386 |
| OPM | 19 | 34 | 52 | 25 | 67 | 91 | 26 | 101 | 126 | 21 | 150 | 170 | 23 | 187 | 209 | 22 | 330 | 251 | 24 | 363 | 382 |
| PDBTM | 18 | 35 | 52 | 17 | 68 | 84 | 15 | 109 | 123 | 15 | 152 | 166 | 16 | 191 | 206 | 14 | 334 | 347 | 15 | 368 | 382 |
| Uniprot | 23 | 33 | 55 | 23 | 66 | 88 | 22 | 105 | 126 | 21 | 150 | 170 | 25 | 188 | 212 | 22 | 330 | 351 | 22 | 367 | 388 |
| TMHMM | 23 | 32 | 54 | 23 | 67 | 89 | 23 | 104 | 126 | 23 | 150 | 172 | 23 | 192 | 214 | 23 | 331 | 353 | 23 | 368 | 390 |
</figtable>
<figtable id="tab:3pbl_TM"> Visualizations for the human D(3) dopamine receptor as a transmembrane protein. Note: Cytoplasmic side of the membrane is different in the pictures
 Human D(3) dopamine receptor as depicted in OPM database (Cytoplasmic side on the bottom) (source) |
 Human D(3) dopamine receptor as shown in PDBTM database (Cytoplasmic side above) (source) |

</figtable>
Voltage-gated potassium channel (Q9YDF8)
<figtable id="tab:1orq_TM_PostProb"> Posterior probability plots Polyphobius and TMHMM for Q9YDF8
 Voltage-gated potassium channel, plot of posterior probabilities of features predicted by Polyphobius |
 Voltage-gated potassium channel, plot of posterior probabilities of features predicted by TMHMM |
</figtable>
| organism: | Aeropyrum pernix (strain ATCC 700893 / DSM 11879 / JCM 9820 / NBRC 100138 / K1) |
|---|---|
| pdb-id: | 1orq (smaller resolution than 2AOL, more residues determined) |
The voltage-gated potassium channel is a tetrameric protein, that causes a voltage-dependent potassium ion permeability of the membrane. It may occur in an opened or closed conformation. This state can be changed by the voltage difference across the membrane.
(source)
To fulfill this function, the protein needs the transmembrane helices. Of these, 7 are predicted according to the Polyphobius output file, but referring to OPM, only three helices should be predicted, according to PDBTM 4 helices are considered true. Looking carefully at the probability plot of Polyphobius in <xr id="tab:1orq_TM_PostProb"/>, one may see, that there actually are only four peaks of transmembrane helix posterior probability, but three of the less higher peaks are considered true as well.
Only 2 of all 7 helices are common among all three methods (see <xr id="tab:1orq_TM" />, picture 3), the third and fourth transmembrane helices are even only predicted by Polyphobius, Uniprot and TMHMM and only Polyphobius and Uniprot, respectively. If not predicted or shown by one of the methods, the helix is considered as membrane loop.
Here OPM has the smallest mean length of transmembrane helices (see <xr id="tab:1orq_TM" />, picture 3). Start and end points of the helices seem to be differing more than in the first described picture (see <xr id="tab:1orq_start_end" />, picture 3), although Polyphobius, Uniprot and TMHMM are quite similar. Personally, considering the features predicted by Polyphobius and TMHMM (see <xr id="tab:1orq_TM_PostProb" />), I would trust the PDBTM version the most in this case. But since in this case, the advantage of Polyphobius to include homologue data is not given, because the BLAST search did not find any homologue sequences, it might on the other hand be a false prediction. This might, to a certain degree, explain the rather diverse result.
<figtable id="tab:1orq_start_end"> Start and end points of each transmembrane helix predicted
| TMH number | 1 | 2 | 3 | 4 | 5 | 6 | 7 | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Method | Length | Start | End | Length | Start | End | Length | Start | End | Length | Start | End | Length | Start | End | Length | Start | End | Length | Start | End |
| Polyphobius | 19 | 42 | 60 | 21 | 68 | 88 | 22 | 108 | 129 | 21 | 137 | 157 | 22 | 163 | 184 | 18 | 196 | 213 | 21 | 224 | 244 |
| OPM | 0 | - | - | 0 | - | - | 0 | - | - | 0 | - | - | 20 | 153 | 172 | 13 | 183 | 195 | 19 | 207 | 225 |
| PDBTM | 32 | 21 | 52 | 24 | 57 | 80 | 0 | - | - | 0 | - | - | 21 | 151 | 171 | 28 | 209 | 236 | 0 | - | - |
| Uniprot | 25 | 39 | 63 | 25 | 68 | 92 | 17 | 109 | 125 | 21 | 137 | 157 | 25 | 160 | 184 | 13 | 196 | 208 | 32 | 222 | 253 |
| TMHMM | 23 | 39 | 61 | 20 | 68 | 87 | 23 | 107 | 129 | 0 | - | - | 23 | 162 | 184 | 20 | 199 | 218 | 20 | 225 | 244 |
</figtable>
<figtable id="tab:1orq_TM"> Visualizations for the Voltage-gated potassium channel as a transmembrane protein. Note: Cytoplasmic side of the membrane is different in the pictures
 Voltage-gated potassium channel as depicted in OPM database (Cytoplasmic side on the bottom) (source) |
 Voltage-gated potassium channel as shown in PDBTM database (Cytoplasmic side above) (source) |

</figtable>
Aquaporin-4 (P47863)
<figtable id="tab:2d57_TM_PostProb"> Posterior probability plots Polyphobius and TMHMM for Q9YDF8
 Aquaporin-4, plot of posterior probabilities of features predicted by Polyphobius |
 Aquaporin-4, plot of posterior probabilities of features predicted by TMHMM |
</figtable>
| organism: | Rattus norvegicus (Rat) |
|---|---|
| pdb-id: | 2d57 (non-mutant) |
Another homotetramer, the water-specific channel Aquaporin-4, has been analyzed. In the first two pictures in <xr id="tab:2d57_TM"/>, the protein is depicted in its tetrameric structure, while in this exercise we only examine one of the four chains. Here Polyphobius again predicted the number of transmembrane helices correctly according to PDBTM and all the other ressources(6 - see Polyphobius output file), while OPM alone shows 8 TMH structures. Taking a closer look at the results, we see that the 2 "missing" TMHs are interpreted as membrane loop in PDBTM (depicted in orange in <xr id="tab:2d57_TM"/>, picture 2) and Polyphobius, which might be due to the short length of the helical structure (10 residues each - see <xr id="tab:2d57_TM"/>). Considering the posterior probability, I would rather prefer one of the 6 helical models.
The 6 shared helices' start and end points are again quite conform among Polyphobius and OPM, PDBTM, Uniprot and TMHMM. The mean length is again around 23, except for the two databases which have a mean length of around 18.
<figtable id="tab:2d57_start_end"> Start and end points of each transmembrane helix predicted
| TMH number | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Method | Length | Start | End | Length | Start | End | Length | Start | End | Length | Start | End | Length | Start | End | Length | Start | End | Length | Start | End | Length | Start | End |
| Polyphobius | 25 | 34 | 58 | 22 | 70 | 91 | 0 | - | - | 22 | 115 | 136 | 22 | 156 | 177 | 21 | 188 | 208 | 0 | - | - | 22 | 231 | 252 |
| OPM | 23 | 34 | 56 | 19 | 70 | 88 | 10 | 98 | 107 | 25 | 112 | 136 | 23 | 156 | 178 | 15 | 189 | 203 | 10 | 214 | 223 | 22 | 231 | 252 |
| PDBTM | 17 | 39 | 55 | 18 | 72 | 89 | 0 | - | - | 18 | 116 | 133 | 20 | 158 | 177 | 18 | 188 | 205 | 0 | - | - | 18 | 231 | 248 |
| Uniprot | 21 | 37 | 57 | 21 | 65 | 85 | 0 | - | - | 21 | 116 | 136 | 21 | 156 | 176 | 21 | 185 | 205 | 0 | - | - | 21 | 232 | 252 |
| TMHMM | 23 | 33 | 55 | 23 | 70 | 92 | 0 | - | - | 23 | 112 | 134 | 23 | 154 | 176 | 23 | 189 | 211 | 0 | - | - | 23 | 231 | 253 |
</figtable>
<figtable id="tab:2d57_TM"> Visualizations for the Aquaporin-4 as a transmembrane protein. Note: Cytoplasmic side of the membrane is different in the pictures
 Rat's Aquaporin-4 as depicted in OPM database (Cytoplasmic side on the bottom) (source) |
 Rat's Aquaporin-4 as shown in PDBTM database (Cytoplasmic side above)(source) |

</figtable>
Comparison Polyphobius, OPM, PDBTM
<figure id="fig:bitshift">

</figure>
In <xr id="fig:bitshift" /> the predicted transmembrane helices of Polyphobius have been used as basis and the start and end point of each TMH in the models of OPM, PDBTM, Uniprot and TMHMM (see <xr id="tab:3pbl_start_end" />, <xr id="tab:1orq_start_end" /> and <xr id="tab:2d57_start_end" />) have been compared to it. The average of each model has been calculated for each helix Polyphobius and OPM, respectively each of the other models, shared. It becomes obvious, that the transmembrane helices of OPM tend to be shifted to the left, compared to the Polyphobius helices, while the PDBTM helices usually are nested inside the Polyphobius membranes due to later starting and earlier end points. The great strength of Polyphobius becomes apparent, when examining the result for 1orq, where no homologue data could be found. (see Voltage-gated potassium channel ). Here all models are very different from Polyphobius' prediction, except for the Uniprot prediction. Taking a closer look, we found out, that the Uniprot prediction among few other methods relies on the Phobius and TMHMM algorithms (see)
Since on average, the hydrophobic belt in a membrane is about 3.5 to 5nm wide and a helix needs approximately 15 to 20 amino acids to span this width, mean lengths of 18 to 23 aas seem reasonable. Nonetheless, especially TMHMM, which seems to predict helices of length 23 in almost all cases, and also Polyphobius and Uniprot tend to have longer helices than that. This fact might be either due to the type of membrane the proteins are located at, or a bias in the algorithms.
What also stood out, was the fact that the cutoff for still counting the structure as an helical structure in the OPM database is really low, considering that an α-helix on average needs 3.6 amino-acid residues per turn.
Contrariwise OPM predicts the least number of transmembrane helices in the Voltage-gated potassium channel protein, which cannot be due to the length of the predicted structure.
Summing up, the method to prefer depends on what demand you have in your result. If you want to make sure your predicted helices are transmembrane helices for sure, you want to choose PDBTM's models, since in our example, they did never assign the label TMH to a structure as the only method. But then, if you want to cover all possible structures, you should choose Polyphobius, since there has not been any transmembrane helix it did not at all predict, considering the two databases.
Signal peptides
Prediction of the presence and location of signal peptide cleavage sites in amino acid sequences.
α-Galactosidase A (P06280)
<figure id="fig:1r46_SP">

</figure>
| organism: | Homo sapiens (Human) |
|---|---|
| pdb-id: | 1r46 |
| Signal-peptide: | 1-31 |
| Signal-peptide sequence: | MQLRNPELHLGCALALRFLALVSWDIPGARA |
As already described in section Transmembrane helices the human α-Galactosidase A does not have transmembrane helical structures and from our background knowledge we know that the protein has a 31 residues long signal peptide at the N-terminus. The protein's pdb structure is available only without signal peptide, since those residues are cleaved off. Therefore no picture is provided here.
In <xr id="fig:1r46_SP"/> the signal peptide prediction of SignalP 4.0 is shown. The cleaved site is clearly indicated after residue 31, since the S-score (green) drops and the C- (red) and Y-score (blue) not only have a peak at this position, but attain their maximum at this point (see SignalP output file )
Serum Albumin (P02768)
| organism: | Homo sapiens (Human) |
|---|---|
| pdb-id: | 1ao6 |
| Signal-peptide: | 1-18 |
| Signal-peptide sequence: | MKWVTFISLLFLFSSAYS |
For this protein an 18 residues long signal peptide is predicted (see SignalP output file) and also reported in various sources (e.g. peptide website, Uniprot). The signal peptide of each chain of the homodimeric protein is shown in <xr id="tab:1ao6_SP"/> in the last picture.
According to the Polyphobius prediction, no transmembrane helix is present, but the signal peptide is also predicted. Those findings underlines the truth of the predicted signal peptide.
The rather high hydrophobicity (see <xr id="tab:1ao6_SP"/>, Figure 1) indicates an export of the protein to another cellular compartment. Examining the Uniprot website, we found that Serum Albumin is secreted into the cell surroundings, for this a membrane needs to be passed and the high hydrophobicity is needed.
<figtable id="tab:1ao6_SP"> Visualizations for the human Serum Albumin protein's signal peptide prediction.
 Human Serum Albumin hydropathy plot (source) |
 Human Serum Albumin plot of scores for signal peptide cleavage site (source) |

</figtable>
Lysosome-associated membrane glycoprotein 1 (P11279)
| organism: | Homo sapiens (Human) |
|---|---|
| pdb-id: | NA |
| Signal-peptide: | 1-28 |
| Signal-peptide sequence: | MAAPGSARRPLLLLLLLLLLGLMHCASA |
<figtable id="tab:P11279_SP"> Visualizations for the human Lysosome-associated membrane glycoprotein's signal peptide prediction.
 Human Lysosome-associated membrane glycoprotein 1 hydropathy plot (source) |
 Human Lysosome-associated membrane glycoprotein 1 plot of scores for signal peptide cleavage site (source) |
</figtable>
As the name implies, LAMP1 has a transmembrane helix (see Polyphobius prediction) and is associated to the plasma membrane, where it presents carbohydrate ligands to selectins. Moreover, the protein shuttles between lysosomes, endosomes, and the plasma membrane ([www.uniprot.org/uniprot/P11279 source]) and thus needs its signal peptide (residues 1-28), which has been predicted by Polyphobius and SignalP (see output). In <xr id="tab:P11279_SP"/> the hydropathy plot and a plot of the different scores used for prediction by SignalP are shown. Again, they underline the predictions of both programs.
Unfortunately, no pdb structure is available.
Aquaporin-4 (P47863)
<figure id="fig:2d57_SP">

</figure>
| organism: | Rattus norvegicus (Rat) |
|---|---|
| pdb-id: | 2d57 |
| Signal-peptide: | none |
| Signal-peptide sequence: | --- |
In <xr id="fig:2d57_SP"/> only a small peak of the C-score at position 41 can be observed, which by far does not reach the threshold of 0.5 for being predicted a cleavage site. All other scores are constantly low, thus no signal peptide cleavage site is predicted. As already mentioned in section Transmembrane helices, Polyphobius correctly predicts no signal peptide, but 6 transmembrane helices. This predicted (and observed) structure is in conformity with the function of a water-specific channel the porin fulfills.
GO terms
QuickGO
<figtable id="tab:QuickGO"> Results of the QuickGO search
| Code | Name |
|---|---|
| GO:0052692 | raffinose alpha-galactosidase activity |
| GO:0051001 | negative regulation of nitric-oxide synthase activity |
| GO:0046479 | glycosphingolipid catabolic process |
| GO:0046477 | glycosylceramide catabolic process |
| GO:0045019 | negative regulation of nitric oxide biosynthetic process |
| GO:0044281 | small molecule metabolic process |
| GO:0043202 | lysosomal lumen |
| GO:0043169 | cation binding |
| GO:0042803 | protein homodimerization activity |
| GO:0016936 | galactoside binding |
| GO:0016798 | hydrolase activity, acting on glycosyl bonds |
| GO:0016787 | hydrolase activity |
| GO:0016139 | glycoside catabolic process |
| GO:0009311 | oligosaccharide metabolic process |
| GO:0008152 | metabolic process |
| GO:0006687 | glycosphingolipid metabolic process |
| GO:0006665 | sphingolipid metabolic process |
| GO:0005975 | carbohydrate metabolic process |
| GO:0005794 | Golgi apparatus |
| GO:0005764 | lysosome |
| GO:0005737 | cytoplasm |
| GO:0005625 | soluble fraction |
| GO:0005576 | extracellular region |
| GO:0005515 | protein binding |
| GO:0005102 | receptor binding |
| GO:0004557 | alpha-galactosidase activity |
| GO:0004553 | hydrolase activity, hydrolyzing O-glycosyl compounds |
| GO:0003824 | catalytic activity |
</figtable>
Since we used QuickGO in Task2 to download the GO terms we decided to refine our GO analysis now. The QuickGO search revealed 28 distinct GO terms (see <xr id="tab:QuickGO"/>). A lot of these are about hydrolase activity and metabolic processes (especially glycosphingolipid biosynthesis), as well as the location of the protein (lysosome) and its enzymatic function. The rather large number of resulting GO terms, may be explained by the listing of all possible terms, especially the quite general ones.
GOPET
<figtable id="tab:GOPET"> Results of the GOPET search
| Result for GOPET search | |||||
|---|---|---|---|---|---|
| GOid | Aspect | Confidence | GO term | ||
| GO:0016798 | Molecular Function Ontology (F) | 98% | hydrolase activity acting on glycosyl bonds | ||
| GO:0004553 | Molecular Function Ontology (F) | 98% | hydrolase activity hydrolyzing O-glycosyl compounds | ||
| GO:0016787 | Molecular Function Ontology (F) | 97% | hydrolase activity | ||
| GO:0004557 | Molecular Function Ontology (F) | 96% | alpha-galactosidase activity | ||
| GO:0008456 | Molecular Function Ontology (F) | 89% | alpha-N-acetylgalactosaminidase activity | ||
</figtable>
Searching the GOPET annotation tool with the AGAL_HUMAN sequence revealed 5 GOIds, which are displayed in <xr id="tab:GOPET"/>. Even maximizing the "maximum number of GO prediction to be displayed per sequence" and minimizing the "confidence threshold for prediction" did not result in any more sequences.
On a first glance, since we already know the name and function of the protein, it is a bit surprising, that alpha-galactosidase activity is only the third entry with 96% confidence. In our already carried out information gathering we learned that α-galactosidase A is a hydrolase thus the first three entries were not surprising. Considering that our enzyme mainly is a glycosidase, the both entries on top of the list make perfekt sense.
Again a bit surprising was the last entry. α-N-Acetylgalactosaminidase is actually used for enzyme replacement therapy, which we mention on our main page. The structure of both enzymes is similar to each other, but this still does not explain the association of this GO term to the AGAL protein. Also, the QuickGO search did not bring up this GO term.
ProtFun2.2
In the output file of ProtFun, the "=>" indicates, which of the subcategories of each category is predicted to be true for the submitted sequence. This prediction is performed on basis of the highest information content. In the pictures below the probabilty, odds and information content (probabilty*odds) are shown separately for each category. The left y-axis is assigned to the probabilty (blue) and the information content (red line), the right y-axis is assigned to the probabilty, which is multiplied by 10 for a better perceptibility.
According to the information content the most likely functional category of the human α-galactosidase A protein is "Cell envelope". Considering our own researches, we would not come to this conclusion, but rather assign a metabolic or regulatory class, since to us known GO terms are for example "glycoside catabolic process", "negative regulation of nitric oxide biosynthetic process" and "small molecule metabolic process".
The prediction that the submitted sequence belongs to an enzyme is right.
In our opinion, it is not always the best evaluation method to decide on the basis of the information content, since the chosen enzyme class "Ligase" indeed has the highest information content, but not the highest probabilty and is, considering literature, wrong. AGAL_HUMAN clearly is a Hydrolase, which is also indicated by the high probabilty. The assigned EC number of the galactosidase is EC=3.2.1.22.
ProtFun does not predict a Gene Ontology category, since the category with the highest information content has odds lower than 1. We actually expected this category to be very unclear since the protein has very diverse cellular functions.
- Probability, Odds and Information Content of the ProtFun2.2 predictions

Functional category
Enzyme class

Gene Ontology category



ProtFun uses a lot of other ressources to predict the probabilties and odds of the categories. Some of the outcomes are listed in <xr id="tab:ProtFun"/>. It is known that the α-galactosidase A protein has a 31 residues long signal peptide on position 1 to 31 which later is cleaved of. Thus the prediction of SignalP 3.0 is right.
The prediction of a propeptide cleavage site could not be confirmed in the human AGAL protein.
ProtFun infered 22 phosphorylation sites at the positions 62, 102, 201, 235, 238, 241, 276, 304, 364, 371, 405, 424, 366, 400, 86, 134, 151, 152, 173, 184, 207, 216. Phosphosite confirms phosphorylation modificatopns at S23, Y134 and H186 of which only one was predicted by NetPhos.
Uniprot claims that at the positions 139, 192, 215 and 408 there are N-linked Glycosilated sites (the last one is only a potential site). All 4 are predicted by NetNGlyc. As predicted by NetOGlyc, there are no (known) O-glycosylated sites.
See also sections Transmembrane helices and Signal peptides for further information on these two topics.
<figtable id="tab:ProtFun"> Output rendered by the individual features used by ProtFun 2.2
| Feature | Output summary | Details |
|---|---|---|
| SignalP 3.0 | Most likely cleavage site between pos. 31 and 32: ARA-LD | Using neural networks (NN) and hidden Markov models (HMM) trained on eukaryotes |
| ProP 1.0 | 1 propeptide cleavage site predicted at position: 196 | Furin-type cleavage site prediction (Arginine/Lysine residues) |
| TargetP 1.1 | No high confidence targeting predition | - |
| NetPhos 2.0 | 22 putative phosphorylation sites | phosphorylation site prediction |
| NetOGlyc 3.1 | No O-glycosylated sites predicted | - |
| NetNGlyc 1.0 | 4 putative N-glycosylated sites at positions 139 192 215 408 | - |
| TMHMM 2.0 | No TM helices predicted | - |
</figtable>
Pfam
The Pfam sequence search revealed one significant Pfam-A match, which is shown in <xr id="fig:pfam_fam"/>.
<figtable id="fig:pfam_fam"> Pfam-A match
| Family | Description | Entry xtype | Clan | Envelope Start | Envelope End | Alignment Start | Alignment End | HMM From | HMM To | Bit score | E-value |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Melibiase | Melibiase | Family | CL0058 | 33 | 149 | 40 | 146 | 41 | 140 | 50.0 | 1.5e-13 |
</figtable>
<figure id="fig:AGAL">

</figure>
The alpha-galactosidase A protein is, according to PFAM, in the family of the Melibiases (see <xr id="tab:Pfam_Melibiose"/>).
The Pfam PF02065 family itself is refered to as "Glycoside hydrolase family 27" and "Glycoside hydrolase family 36", the AGAL_HUMAN falls into the first category. Their common characteristic is that the members of this family are glycoside hydrolases (EC 3.2.1.). The AGAL enzyme catalyzes the hydrolisis of the disaccharide Melibiose (D-Gal-α(1→6)-D-Glc) into its two components galactose and glucose.
<figtable id="tab:Pfam_Melibiose"> Melibiase Identifiers
| Melibiase Identifiers | |||||
|---|---|---|---|---|---|
| Symbol | Melibiase | ||||
| Pfam | PF02065 | ||||
| Pfam clan | CL0058 | ||||
| InterPro | IPR000111 | ||||
| SCOP | 1ktc | ||||
| SUPERFAMILY | 1ktc | ||||
| CAZy | GH27 | ||||
</figtable>
The alignment of positions 40 - 146 of the AGAL_HUMAN protein sequence and the matching HMM used in this prediction, is shown in <xr id="fig:pfam_ali" />. According to the color code, indicating the degree of confidence of each aligned position, there is an overall very good agreement of the Hidden Markov Model and the sequence, except for the residues 17-46. Checking our background knowledge and the Uniprot database we could not find a very interesting or abnormal region here, but the signal peptide cleavage site and a beta strand at position 42-46.
Our query protein belongs to a rather large Clan, the Glyco_hydro_tim Clan (CL0058), which includes 4 CAZy-Clans (GH-A, GH-D, GH-H and GH-K). They main attribute of all the included glycosyl hydrolase enzymes is the hold of a a TIM barrel fold (eight α-helices and eight parallel β-strands that alternate along the peptide backbone source). This fold (residue 31 - 324) can be well seen in <xr id="fig:AGAL"/>, where the 3D structure of α-Galactosidase A is depicted.
The InterPro protein sequence analysis and classification assigns the enzyme to the "IPR000111 Glycoside hydrolase, clan GH-D" family. As mentioned before, GH-D is part of the Glyco_hydro_tim Clan and thus it is not surprising, that the description is almost the same. According to InterPro, there are 6 IPR000111 family members in the human body.
The Structural Classification of Proteins (SCOP) finds two domains, an Amylase, catalytic domain (c.1.8.1, residues 32 - 323) and an alpha-Amylases, C-terminal beta-sheet domain (b.71.1.1, residues 324 - 421). The catalytic domain is an alpha and beta protein (a/b) with TIM beta/alpha-barrel fold, which is consonant with the affiliation to the Glyco_hydro_tim Clan. The C-terminal beta-sheet domain is an all beta protein, the fold and superfamily is Glycosyl hydrolase domain. This is also not surprising to us.
AGAL is in the CAZy GH27 (Glycoside Hydrolase Family 27) family, together with a-N-acetylgalactosaminidase (EC 3.2.1.49; as mentioned before, used for enzyme replacement therapy), isomalto-dextranase(EC 3.2.1.94) and b-L-arabinopyranosidase (EC 3.2.1.88). See http://www.cazypedia.org/index.php/Glycoside_Hydrolase_Family_27 TODO
<figure id="fig:pfam_ali">

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
</figure>
Other programs and ressources
A simple internet search for "protein sequence prediction" reveals a confusingly high number of different ressources and databases. Clicking on the first entry leads to Rostlab's Predict protein. This program seems to predict nearly anything:
- Multiple sequence alignment
- ProSite sequence motifs
- low-complexity retions (SEG)
- Nuclear localisation signals
- and predictions of
- secondary structure
- solvent accessibility
- globular regions
- transmembrane helices
- coiled-coil regions
- structural switch regions
- B-value
- disordered regions
- intra-residue contacts
- protein protein and protein/DNA binding sites
- sub-cellular localization
- domain assignment
- beta barrels
- cysteine predictions and disulphide bridges
Performing a search reveals that the tool gives you an overview of all the above mentioned features. On a first glance it seemed rather confusing, due to the vast amount of informations provided. Anyhow I believe giving the output some more attentiveness, it is a good tool to gather a lot of information in one place. A very good feature is that the output is presented in many different ways which can be switched at any time. A quick look at the properties we examined in the task, shows, that Predict Protein probably does not distinguish very well between signal peptides and transmembrane helices, since for the α-Galactosidase A it predicts a TM, where there actually is signal peptide (position 11-28; see section Transmembrane helices and Signal peptides)
Searching the same term again at the NCBI homepage (see search) reveals a huge mass of reviews on that topic, mainly on predicting secondary structure and function out of the protein sequence, but also on less common topics like predicting the protein solubility (see here and here) , or more recently emerged whole genome sequence approaches (see).
A really different topic was brought up by Smialowski P, Martin-Galiano AJ, Cox J and Frishman D. who give an overview of techniques to predict experimental properties of proteins from sequence (see) and thus predict experimental success in cloning, expression, soluble expression, purification and crystallization
Summarizing, it is very hard to get along in the vast amount of prediction tools and methods and one can spend a lot of time on finding out which one fits best to the requirements of one's topic.