Metachromatic leukodystrophy
Contents
Summary
Metachromatic leukodystrophy (MLD) is a genetic disorder that affects nerves, muscles, other organs, and behavior. It slowly gets worse over time.
MLD is a lysosomal storage disease that is caused by a deficiency in Arylsulfatase A, leading to an accumulation of the sulfolipid sulfatide . This affects the growth and/or development of myelin. The myelin covering acts as an insulator around nerve fibers throughout the central and peripheral nervous systems.
Phenotype
The disease leads to a toxic buildup of lipids in cells in the nervous system, liver, and kidneys (see below for mechanism). This impairs the function of the cells of the brain and spinal cord as well as the nerves connecting the brain and spinal cord to muscles and sensory cells. This affects the muscle tone, leading to abnormally high muscle tone as well as decreased muscle tone. This in turn leads to walking and feeding difficulties, frequent falls, incontinence, speech difficulties with slurring, etc. It can also effect nerve function and lead to decreased mental function, personality changes, etc. (see PubMed Health).
There are three forms of MLD: late infantile, juvenile, and adult. (see e.g. National Institute of Neurological Disorders and Stroke).
- The late infantile form is most common (50 to 60 per cent of patients). It sets in between 12 and 20 months following birth. Affected children have difficulty walking after the first year of life and subsequently show muscle wasting leading to paralysis, loss of vision leading to blindness, and dementia. Most children with this form of MLD die by age 5.
- The juvenile form sets in between 3-10 years of age. It usually begins with impaired school performance. Subsequently symptoms similar to the late infantile form occur, but with slower progression. Death occurs 10 to 20 years following the onset of the disease.
- The adult form commonly begins after age 16. It is first seen as a psychiatric disorder or progressive dementia. Symptoms include impaired concentration, ataxia, seizures, dementia, and tremor. Death occurs 20 (or 6 according to other sources) to 30 years following the onset of the disease.
Metachromatic leukodystrophy gets its name from the microscopic appearance of cells with the sulfatide (lipid) accumulation that occurs in this disorder. The sulfatides form granules which are metachromatic, which means they pick up color differently than surrounding cellular material when stained for microscopic examination.
Cross-references
See also description of this disease in
- Wikipedia
- OMIM
- HGMD
- PubMed Health
- GeneReviews
- National Institute of Neurological Disorders and Stroke
- MedMerits
Biochemical disease mechanism
- The symptoms of MLD are caused by the gradual degradation of the myelin sheaths that act as insulators around nerve fibers throughout the central and peripheral nervous systems.
- The reason for the degradation is the accumulation of sulfatides (sulfated glycosphingolipids), which gradually destroys the cells forming the myelin sheath.
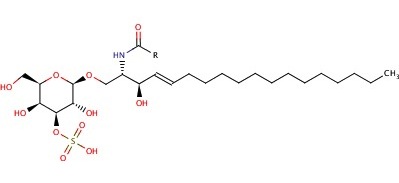 galactosylceramide sulfate; source: http://www.ebi.ac.uk/chebi
galactosylceramide sulfate; source: http://www.ebi.ac.uk/chebi - The sulfatides accumulate in the lysosome when the enzyme Arylsulfatase A responsible for breaking up sulfatides, is not working. Hence the disease is called a "lysosomal storage disease".
- The protein Arylsulfatase A
- has a sequence length of 507 aa,
- is located in the lysosome,
- has a Ca cofactor
- is encoded by the Gene ARSA (see #Genomic information) on chromosome 22.
- In cells from patients with juvenile and adult forms of MLD, von Figura et al. (1983) found severe deficiency in the arylsulfatase polypeptide, because the mutant enzyme was rapidly degraded upon transport into lysosomes.
- Some patients with clinical metachromatic leukodystrophy have normal arylsulfatase A activity but lack an activator protein that is involved in sulfatide degradation. (These pages will focus only on mutations in the original Aryslulfatase A.)
- The fold of the ARS A monomer has significant structural analogy to another hydrolytic enzyme, the alkaline phosphatase. (The active centers are located in largely identical positions.) The functionally essential formylglycine is located in a positively charged pocket and acts as ligand to an octahedrally coordinated metal ion. In the proposed catalytic mechanism, the aldehyde accepts a water molecule to form a hydrate. One of the two hydroxyl groups hydrolyzes the substrate sulfate ester via a transesterification step, resulting in a covalent intermediate. The second hydroxyl serves to eliminate sulfate under inversion of configuration through C-O cleavage and reformation of the aldehyde. (see mechanism graphics in publication based on the first crystal structure)



{kind=link}
Cross-references
- sulfatide entry at ChEBI
- sulfatide entry in KEGG
- MLD entry in KEGG
- lysosome pathway in KEGG
- sphingolipid metabolism in KEGG
- KEGG reactions catalysed by ARS A
- MetaCyc reaction of ARS A
- Sphingolipid metabolism in NCBI BioSystems
- Interactions in STRING
- ARS A in Uniprot
- ARS A in Nextprot
Treatment
- Bone marrow and umbilical cord blood transplantation to replace the defective enzyme may slow down the progression.
- Gene therapy is under development as a possible solution to correct the underlying genetic abnormality.
- In Europe, ongoing clinical trials are evaluating the safety and efficacy of a recombinant human arylsulfatase A (rhARSA) enzyme, metazym.
- Physical, occupational and speech therapy to maintain the range of motion.
References
Genomic information
The gene for ARS A lies on chromosome 22.

MLD shows autosomal recessive inheritance. Only the ASA-/ASA- genotype was is associated with the development of both early- and late-onset MLD, including neuropsychiatric symptoms. Variations in the phenotype can be attributed to different mutations of the ARSA gene that affect the protein activity differently.
Cross-references
Reference sequence, mutations and sequence features
The Uniprot entry for ARS A P15289 gives the protein sequence used as reference in this practical:
>sp|P15289|ARSA_HUMAN Arylsulfatase A OS=Homo sapiens GN=ARSA PE=1 SV=3 MGAPRSLLLALAAGLAVARPPNIVLIFADDLGYGDLGCYGHPSSTTPNLDQLAAGGLRFT DFYVPVSLCTPSRAALLTGRLPVRMGMYPGVLVPSSRGGLPLEEVTVAEVLAARGYLTGM AGKWHLGVGPEGAFLPPHQGFHRFLGIPYSHDQGPCQNLTCFPPATPCDGGCDQGLVPIP LLANLSVEAQPPWLPGLEARYMAFAHDLMADAQRQDRPFFLYYASHHTHYPQFSGQSFAE RSGRGPFGDSLMELDAAVGTLMTAIGDLGLLEETLVIFTADNGPETMRMSRGGCSGLLRC GKGTTYEGGVREPALAFWPGHIAPGVTHELASSLDLLPTLAALAGAPLPNVTLDGFDLSP LLLGTGKSPRQSLFFYPSYPDEVRGVFAVRTGKYKAHFFTQGSAHSDTTADPACHASSSL TAHEPPLLYDLSKDPGENYNLLGGVAGATPEVLQALKQLQLLKAQLDAAVTFGPSQVARG EDPALQICCHPGCTPRPACCHCPDPHA
Human lysosomal arylsulfatase A (ARS A) requires the posttranslational oxidation of the -CH2SH group of a conserved cysteine to an aldehyde, yielding a formylglycine. Without this modification sulfatases are catalytically inactive (one cause of the disease!).
ARS A exists both as a single chain of 58 kDa (component A) or as a chain of 50 kDa (component B) linked by disulfide bond(s) to a 7 kDa chain (component C).
ARS A binds a Ca-ion that is important for function.