Gaucher Disease: Task 04 - Structural Alignment
<css>
table.colBasic2 { margin-left: auto; margin-right: auto; border: 1px solid black; border-collapse:collapse; }
.colBasic2 th,td { padding: 3px; border: 1px solid black; }
.colBasic2 td { text-align:left; }
/* for orange try #ff7f00 and #ffaa56 for blue try #005fbf and #aad4ff
maria's style blue: #adceff grey: #efefef
- /
.colBasic2 tr th { background-color:#efefef; color: black;} .colBasic2 tr:first-child th { background-color:#adceff; color:black;}
</css>
Contents
Exploring Structural Alignments
Used data set
<figtable id="data_set">
| Sequence Set | ||||
|---|---|---|---|---|
| PDB ID | Protein name | CATH Superfamily | Category | Seq. ID% to reference |
| 1OGS | Glucocerebrosidase | Glycosidases (3.20.20.80) | unfilled binding sites (reference structure) | reference str. |
| 2XWD | Glucocerebrosidase | Glycosidases (3.20.20.80) | filled binding sites | 99 |
| 2NSX | Glucocerebrosidase | Glycosidases (3.20.20.80) | filled binding sites | 100 |
| 2NT1 | Glucocerebrosidase at neutral pH | Glycosidases (3.20.20.80) | Sequence identity >60% | 100 |
| 2F7K | Pyridoxal kinase | Hydroxyethylthiazole kinase-like domain (3.40.1190.20) | unrelated <30% (identical in C) | 3 |
| 2GEP | Sulfite reductase | Adolase class I (3.20.20.70) | identical in CAT | 7 |
| 2ISB | Fumarase of FUM-1 from Archaeoglobus Fulgidus | Fumarase (3.20.130.10) | identical in CA | 10 |
| 2DJF | Human dipeptidyl peptidase I (in complex) | Cysteine proteinases (3.90.70.10) | identical in C | 7 |
| 2QGU | Phospholipid-binding protein from Ralstonia solanacearum (in complex) | Phospholipid-binding protein (1.10.10.640) | different in C | 7 |
</figtable>
Structural alignment methods results
<figtable id="struct_alis">
| PDB ID of second molecule | Pymol | SSAP | LGA | TopMatch | SAP | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| RMSD C-alpha (#atoms) | RMSD all atoms (#atoms) | RMSD all atoms binding site (#atoms) | RMSD (#atoms) | SSAP score | RMSD (#atoms) | LGA score | RMSD/E_r (#atoms/L) | S | S_r | Un-weighted RMSD (#atoms) | SAP score | |
| 2XWD | 0.302 (406) | 0.35 (3032) | 0.172 (35) | 0.89 (492) | 95.39 | 0.75 (490) | 98.277 | 0.75 (490) | 485 | 0.71 | 0.886 (493) | 75730.101562 |
| 2NSX | 0.16 (432) | 0.196 (3383) | 0.110 (30) | 0.24 (497) | 97.37 | 0.23 (497) | 100.000 | 0.23 (497) | 496 | 0.23 | 0.230 (498) | 83186.492188 |
| 2NT1 | 0.226 (454) | 0.251 (3380) | 0.149 (39) | 0.68 (497) | 96.09 | 0.50 (495) | 99.287 | 0.50 (495) | 493 | 0.49 | 0.682 (498) | 76803.125000 |
| 2F7K | 18.823 (165) | 18.947 (1049) | - | 10.85 (177) | 48.29 | 2.92 (93) | 17.684 | 4.22 (91) | 65 | 4.02 | 16.736 (324) | 1945.130493 |
| 2GEP | 21.721 (159) | 21.9 (1006) | [0.002 (2/38)] | 4.85 (231) | 61.57 | 3.10 (60) | 7.709 | 3.07 (55) | 46 | 2.95 | 24.904 (453) | 1557.686279 |
| 2ISB | 12.845 (36) | 14.495 (245) | - | 14.52 (140) | 43.29 | 3.23 (55) | 16.689 | 3.09 (64) | 53 | 3.01 | 22.236 (164) | 697.835266 |
| 2DJF | 16.382 (75) | 16.896 (472) | [0.000 (1/16)] | 9.87 (90) | 41.42 | 2.98 (49) | 25.219 | 2.21 (58) | 53 | 2.11 | 11.958 (102) | 665.702637 |
| 2QGU | 22.773 (93) | 22.457 (592) | 21.03 (156) | 37.57 | 3.35 (34) | 10.973 | 2.55 (43) | 38 | 2.47 | 17.576 (176) | 559.580811 | |
</figtable>
LGA
The best structure alignment was created with 2NSX. LGA does not only align all 497 atoms of both structures, it also results to the best score of 100 and the lowest RMSD (0.23, <xr id="struct_alis"/>). The structures with a high sequence identity show similar character of their alignments. All the RMSDs of these three structure alignments (2NSX, 2XWD, 2NT1) are low. In comparison to the remaining structural alignments they show high scores and a great number of aligned atoms. However, there cannot be seen a direct correlation between RMSD and sequence identity.
LGA shows RMSD values below 4 for all structural alignments (<xr id="struct_alis"/>). On the first sight, it seem to be not a bad alignment, but a closer look at the number of aligned residues shows that only a few aligned residues lead to this value. In general LGA aligns less atoms than other methods. Skipping residues with a greater distance may explains why LGA has no alignments with an RMSD of 20 like other methods (Pymol).
SSAP
The CATH provided method aligns more atoms of two structures than LGA. However, this results to a higher RMSD of each structural alignment. SSAP shows again good values for its similar structures (2NSX, 2XWD, 2NT1)<xr id="struct_alis"/>.
For the structural alignment of both proteins (1OGS, 2GEP) in the same CAT class shows a relative high number of aligned residues (231), although they have a sequence identity of only 7%. Nevertheless, it has a lower RMSD (4.85) than the other alignments which have less aligned residues.
Pymol Visualisation
With Pymol we aligned the reference structure to a structure of our sequence set in <xr id="data_set"/> in two different ways. First, we aligned all atoms of both structures. Second, we focused on the alignment of the C-alpha atoms. In addition, we calculated RMSD only between all atoms within a distance of 6 Å from the ligand, NAG (as described in lab journal of task 05). In all cases the alignments of a structure pair only differ slightly in their composition. Moreover, in all cases the alignment of the C-alpha atoms (and the binding site atoms) has the lower RMSD (see <xr id="struct_alis"/>). The alignments based on C-alpha atoms are shown in <xr id="pymol"/>.
The alignment of 1OGS with 1NSX has the lowest RMSD of 0.16. Most of the C-alpha atoms are aligned. A few deviations between the structures can be observed, however, the differences occur only in loops. The same applies for the structure 2NT1. Both structures have a 100% sequence identity to the reference structure (<xr id="data_set"/>). 2XWD, which has a sequence identity to 1OGS_A of 99%, shows indeed a very similar structure to 1OGS, differs not only in loops but also in the secondary structure. An alpha helix as well as a loop deviate extremely from the reference structure and marked yellow in the image. The RMSD of the binding sites atoms is low for those three structures (<xr id="struct_alis"/>)).
For all other alignments, the aligned structures have nothing structural in common. Even structures that share same CATH levels are miss-aligned in their secondary structures. For structures of the third CATH level (3.20.20 TIM Barrel) at least one aligned helix or beta sheet was expected.
The main reason for that may be the very low sequence identities of these structures to Glucocerebrosidase.
<figure id="pymol" >

aligned to 2XWD, with yellow colored residues that differ more from 1OGS than the remaining residues of 2XWD.

aligned to 2NSX

aligned to 2NT1

aligned to 2F7K

aligned to 2GEP
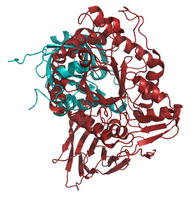
aligned to 2ISB

aligned to 2DJF

aligned to 2QGU








Pymol-Alignment based on C-alpha atoms: 1OGS_A (red) to structures (blue) of the set listed in <xr id="data_set"/>. </figure>
TopMatch
TopMatch aligns only the C-alpha atoms of the structures. For the first three structures, which have a high sequence identity to our protein 1OGS_A, the length of the alignment (L) is high and the RMSD (E_r) is low (below 1). For the remaining five sequences, which have a much lower sequence identity to our protein, the RMSD becomes higher and the number of superposed residues drastically lower.
In <xr id="struct_alis"/> also the S and S_r scores are listed. The similarity score S depends on the error between each of the aligned residues and a scaling factor (sigma). The lower the error for all aligned residues, the higher S. From S, a normalized similarity per residue is calculated, dividing S by L. From s the distance error S_r is calculating (using the sigma). S and S_r are comparable to - but usually lower than - L and E_r, respectively.
SAP
The results for SAP in <xr id="struct_alis"/> show that for the sequences with a high similarity to the target, the number of superposed residues is high and the RMSD is low, similarly to TopMatch. For the sequences with a lower sequence similarity to the target, the RMSD becomes much higher (higher than on TopMatch) and the number of superposed residues lower (but remains higher than in TopMatch). This may be explained, that SAP uses the whole structure for the superposition. SAP score is very high and correlates negatively with the RMSD (the higher the score, the lower the RMSD).
Suggestion
The most satisfying results we got from LGA. The method finds the right balance between aligning as many residues as possible to get a low RMSD. This works not only for proteins with similar structure, but also for different structural compositions. But it is important to consider that always the number of aligned atoms must be taken into account.
For those who want a quick and user friendly overview, we would recommend SSAP of CATH. The results are quite good and easy to understand. No knowledge about the program and its use is needed. Compared to LGA, SSAP is easier but the results are simpler. For LGA you need some time to understand the use and the output but you will get more explicit result.
For a pretty visualisation, it is the best to use Pymol. With Pymol it can be seen which secondary structures are aligned and which are deviating from each other. But the calculated results of Pymol as well as SAP and TopMatch (listed in <xr id="struct_alis"/>) were not compelling in any way.
Evaluation of structural alignments and sequence alignments
In this task, we evaluated HHblits alignments we produced in task 02 using the hhmakemodel modelling tool and the LGA structural alignment tool. For this we applied the script hhmakemodel.pl from the HHblits package to produce very simple models of our query sequence, P04062, from the selected HHblits hits from the PDB by simply copying the C-alpha coordinates of the aligned residues. Then we evaluated how good the alignments are by aligning the models to our reference structure 1OGS_A.
Results
We selected seven PDB hits for modelling with hhmakemodel. This hits were found using an HHblits search (2 iterations against uniprot20 followed by one iteration against pdb_full) with the query sequence P04062. In the first part of <xr id="hhblits_lga_scores"/> HHblits scores of sequence alignments between each hit and the query are presented: probability, E-value, score, alignment length and sequence identity. Using the PDB structures of those hits, we built structural models of the query using hhmakemodel and compared the models to the reference structure 1OGS_A using LGA. In the second part of the table the following LGA scores of structural alignments between each model and 1OGS_A are shown: number of superimposed residues, RMSD, seq_id, LGA_S and LGA_Q.
<figtable id="hhblits_lga_scores">
| HHblits sequence alignments | LGA structural alignments | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| PDB_ID | Probability | E-value | Score | Aligned_cols | Identities(%) | Superimposed residues (N) | Seq_Id(%) | RMSD | LGA_S | LGA_Q |
| 2v3f_A | 100.00 | 2.4e-132 | 1078.26 | 497 | 100 | 492 | 98.98 | 0.77 | 97.938 | 56.371 |
| 2nt0_A | 100.00 | 4.4e-132 | 1074.98 | 496 | 100 | 496 | 99.80 | 0.19 | 99.783 | 173.564 |
| 2wnw_A | 100.00 | 3.7e-107 | 870.15 | 439 | 29 | 430 | 93.49 | 1.58 | 80.275 | 25.550 |
| 3kl0_A | 100.00 | 1.1e-77 | 633.96 | 356 | 19 | 340 | 84.12 | 1.96 | 53.971 | 16.523 |
| 3s2c_A | 98.79 | 7.7e-13 | 139.02 | 246 | 14 | 221 | 87.78 | 2.49 | 29.746 | 8.524 |
| 1fob_A | 97.76 | 1.1e-08 | 101.53 | 197 | 16 | 168 | 85.71 | 2.49 | 21.221 | 6.487 |
| 1ur1_A | 95.65 | 9.8e-05 | 73.33 | 175 | 11 | 140 | 70.71 | 2.75 | 16.681 | 4.906 |
</figtable>
To see whether there is any correlation between model similarity to the reference structure and any of the alignment scores, we calculated Pearson's correlation coefficient between all pairs of the HHblits and LGA scores, which are presented in <xr id="pearsons_cc"/>.
<figtable id="pearsons_cc">
| HHblits | ||||||
|---|---|---|---|---|---|---|
| Aligned_cols | Identities(%) | Probability | E-value | Score | ||
| LGA | Superimposed residues (N) | 0.9999 | 0.8310 | 0.8530 | -0.5429 | 0.9929 |
| Seq_Id(%) | 0.8642 | 0.7958 | 0.8516 | -0.7873 | 0.8218 | |
| RMSD | -0.9372 | -0.9441 | -0.7183 | 0.4593 | -0.9392 | |
| LGA_S | 0.9690 | 0.8389 | 0.8458 | -0.5711 | 0.9383 | |
| LGA_Q | 0.6934 | 0.8304 | 0.4583 | -0.2668 | 0.6978 | |
</figtable>
The number of superposed residues of LGA alignment is strongly positive correlated to the HHblits alignment length and sequence identity, probability and score. Same correlations hold for the LGA alignment sequence identity and LGA_S score, whereas the RMSD shows strong negative correlations to the same HHblits scores. The LGA_Q score is positive correlated with these HHblits scores as well, but significantly strong only with the HHblits sequence identity. The HHblits E-value has the opposite correlation tendencies to the LGA scores than the other HHblits scores. It is strongly negative correlated to the LGA sequence identity, medium negative correlated to the number of superposed residues of LGA alignment and LGA_S, medium positive correlated to the RMSD and slightly negatively correlated to LGA_Q. This means, that HHblits hits with a high alignment length and sequence identity, probability and score lead to good quality of hhmakemodel models (with high number of superposable residues, sequence identity and scores and low RMSD to the reference structure). Low E-value of HHblits hits also contributes to the model quality.