Task 9 (MSUD)
Contents
Results
For this task we have chosen 5 mutations from HGMD and dbSNP. 2 of them are neutral mutations which do not have functional changes over the protein structure. Following are the 5 mutations:
| Database | Accession_code | Mutation | Pathogenic |
| dbSNP | rs11549938 | M82L | False |
| dbSNP | rs141086188 | A222T | False |
| HGMD | CM045934 | C264W | True |
| HGMD | CM062448 | R346H | True |
| dbSNP | rs61736656 | I361V | False |
Selection of structure model
The following table shows an overview of all structures that are available for our protein:
| Entry | Method | Resolution [Å] | Chain | Positions | R-value | pH | gaps |
|---|---|---|---|---|---|---|---|
| 2BFD | X-ray | 1.39 | A | 46-445 | 0.15 | 5.5 | 289GLY-292SER,293THR-313ASP |
| 2BFF | X-ray | 1.46 | A | 46-445 | 0.15 | 5.5 | 301ARG-305GLU |
| 1X7Y | X-ray | 1.57 | A | 46-445 | 0.15 | 5.8 | 287ARG-313ASP |
| 2BFC | X-ray | 1.64 | A | 46-445 | 0.144 | 5.5 | 288ILE-313ASP |
| 2BFE | X-ray | 1.69 | A | 46-445 | 0.15 | 5.5 | 289GLY-291HIS,293THR-313ASP |
| 1X7Z | X-ray | 1.72 | A | 46-445 | 0.154 | 5.8 | 301ARG-306VAL |
| 1X7W | X-ray | 1.73 | A | 46-445 | 0.148 | 5.8 | 287ARG-313ASP |
| 2BFB | X-ray | 1.77 | A | 46-445 | 0.145 | 5.5 | 287ARG-313ASP |
| 2BEW | X-ray | 1.79 | A | 46-445 | 0.147 | 5.5 | 301ARG-307ASN |
| 1V1R | X-ray | 1.80 | A | 46-445 | 0.158 | 5.5 | 222ASN-231SER,286TYR-314HIS |
| 2BEV | X-ray | 1.80 | A | 46-445 | 0.139 | 5.5 | 301ARG-307ASN |
| 1U5B | X-ray | 1.83 | A | 46-445 | 0.156 | 5.8 | 301ARG-306VAL |
| 1WCI | X-ray | 1.84 | A | 46-445 | 0.149 | 5.5 | 301ARG-309TRP |
| 1OLS | X-ray | 1.85 | A | 46-445 | 0.172 | 5.5 | 292SER-295ASP,298SER-300TYR,301ARG-308TYR |
| 2J9F | X-ray | 1.88 | A/C | 46-445 | 0.171 | 5.5 | 300TYR-313ASP |
| 2BEU | X-ray | 1.89 | A | 46-445 | 0.171 | 5.5 | 301ARG-307ASN |
| 1OLU | X-ray | 1.90 | A | 46-445 | 0.161 | 5.5 | 26ASN-30GLY,287ARG-313ASP |
| 1V16 | X-ray | 1.90 | A | 46-445 | 0.132 | 5.5 | 288ILE-313ASP |
| 1V11 | X-ray | 1.95 | A | 46-445 | 0.139 | 5.5 | 288ILE-313ASP |
| 1V1M | X-ray | 2.00 | A | 46-445 | 0.13 | 5.5 | 289GLY-313ASP |
| 1X80 | X-ray | 2.00 | A | 46-445 | 0.161 | 5.8 | 287ARG-313ASP |
| 1X7X | X-ray | 2.10 | A | 46-445 | 0.149 | 5.8 | 287ARG-313ASP |
| 1OLX | X-ray | 2.25 | A | 46-445 | 0.161 | 5.5 | 301ARG-307ASN |
| 1DTW | X-ray | 2.70 | A | 46-445 | 0.224 | 7.5 | 301ARG-314HIS |
Unfortunately all structures contain gaps that span positions 302-304 (corresponding to 347-349 in the reference sequence), so we cannot create a composite structure, that does not contain this gap. We chose 2BFF because it has the smallest gap, a good resolution and a low R-value. It is not resolved at physiological pH, but the only structure with pH 7.5 (1DTW) has a bad resolution. The RMSD between 2BFF and 1DTW is about 0.3, so the different pH does not lead to a different structure and therefore the low pH at that 2BFF was resolved should not be a problem.
Visualization of mutant structures
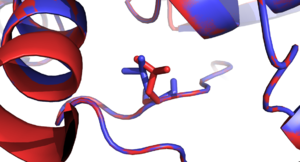
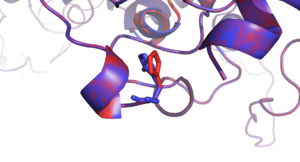
The mutagen tool of PyMOL was used to introduce mutations to the protein structure. Mutations and their neighboring residues are visualized and shown in following figures.
- Visualization of mutations in BCKDHA (PDB: 2BFF, all residue positions refer to the reference sequence of BCKDHA)

Local environment of residue 82. Original methionine is replaced by leucine.

Local environment of residue 222. Original alanine is replaced by threonine.

Local environment of residue 264. Original cysteine is replaced by tryptophan. Clearly, the mutant residue tryptophan clashes with the binding site (Green) for ion binding.

Local environment of residue 346. Original arginine is replaced by histidine.

Local environment of residue 361. Original isoleucine is replaced by valine.





Mutant structures (SCWRL)
Energy comparisons
FoldX
Resulted structural models were compared to the structures calculated by SCWRL. By using alignment tool of PyMOL, we did not find any global deviation between the structures. Sequentially, structures produced by SCWRL have some residues missing. Following table shows the sequential difference.
| Mutation | RMSD(Å) | Missing residues in SCWRL |
| M82L | 0.0 | SER76,HIS96,MET146,LEU325,SER326 |
| A222T | 0.0 | SER76,HIS96,MET146,LEU325,SER326 |
| C264W | 0.0 | SER76,HIS96,MET146,LEU325,SER326 |
| R346H | 0.0 | SER76,HIS96,MET146,LEU325,SER326 |
| I361V | 0.0 | SER76,HIS96,MET146,LEU325,SER326 |
In 3 mutant structures the mutation residues have different side-chain conformation between results of FoldX and SCWRL. The other 2 mutant structures do not show such difference.
- Mutation residues with different side-chain conformations (FoldX in red, SCWRL in blue)